

biokhimia_ekz
.pdf39.Роль анаэробного и аэробного распада глюкозы в мышцах. Судьба молочной кислоты.Глюкоза вступает в процесс гликолиза и в результате образуется 2 ПВК (высвобождается 8 АТФ). Далее ПВК в анаэробных условиях превращается в 2 МК и 2 АТФ. В аэробных условиях происходит окислительное декарбоксилирование ПВК до 2 АцКоА (6 АТФ), который запускает ЦТК, в результате чего образуется СО2, Н2О и 24 АТФ. Судьба молочной кислоты – цикл кори – она образуется в мышцах, эритроцитах и в сетчатке глаза. Молочная кислота является тупиком метаболизма. Если молочной кислоты образуется много – это лактатный ацидоз. Цикл Кори – это соотношение содержания глюкозы и молочной кислоты в различных органах и тканях. Глюкоза из печени поступает в кровь, затем в мышцы, где синтезируется гликоген. Потом он распадается, образуется глюкоза, которая превращается в молочную кислоту. 4/5 молочной кислоты идут в кровь, в печень, превращается в ПВК, который участвует в глюконеогенезе в печени, в результате чего образуется глюкоза и все начинается сначала. Одна молекула глюкозы дает 6 АТФ – процесс самообслуживания. 1/5 молочной кислоты превращается в ПВК, АцКоА, ЦТК, СО2, Н2О, 12 АТФ.
40.Схема аэробного окисления углеводов, образование ПВК, челночные механизмы транспорта водорода.
Три этапа аэробного распада углеводов: 1) гликолиз до ПВК, при этом образуется 8 АТФ 2) окислительное декарбоксилирование ПВК, при этом образуется 6 АТФ 3) АцКоА запускает ЦТК, в итоге образуется 24 АТФ, т.о. на одну молекулу глюкозы приходится 38 молекул АТФ.
Митохондриальная мембрана не проницаема для Н, он транспортируется через челночные механизмы – глицеролфосфатный челночный механизм, Маолат-аспартатная челночная система.
41.Окислительное декарбоксилирование ПВК, связь с дых цепью.
В реакции окислительного декарбоксилирования ПВК образуется 2 АцКоА, 2 СО2 и 2 НАДН, который является источником Н для дыхательной цепи, в результате чего образуется 2 воды и 6 АТФ. Реакцию катализирует мультиферментный комплекс, который включает 5 кофакторов и 3 фермента. Кофакторы: тиаминдифосфат, липоевая кислота, НS-КоА, ФАД, НАД. Ферменты: пируватдегидрогеназа декарбоксилирующая, липоацетилтрансфераза, липоамиддегидрогеназа.
42.ЦТК, связь с дых цепью.
1) Конденсация АцКоА и оксалоацетата, образуется цитрат. 2) превращение цитрата в изоцитрат через цис-аконитовую кислоту. 3) прямое декарбоксилирование изоцитрата, НАДН – источник Н, проступает в дых цепь 3АТФ + Н2О. 4) окислительное декарбоксилирование а-кетоглутората, сложный а-кетоглуторатдегидрогеназный комплекс, включающий 3 фермента и 5 кофакторов (ТДФ, НSКоА, НАД, ФАД, липоевая кислота). НАДН (3АТФ+вода). 5) реакция субстратного фосфорилирования сукцинилКоА, ГТФ (1АТФ). 6) реакция окисления сукцината в фумарат, образуется ФАДН2 (2АТФ и вода). 7) реакция гидратации фумарата с образованием малата. 8) Окисление малата путем дегидрирования, образуется оксалоацетат и НАДН (3АТФ и вода). Суммарное уравнение: АцКоА + 2H2O + Фн + ГДФ 3НАДН + ФАДН2 + ГТФ + НSКоА.
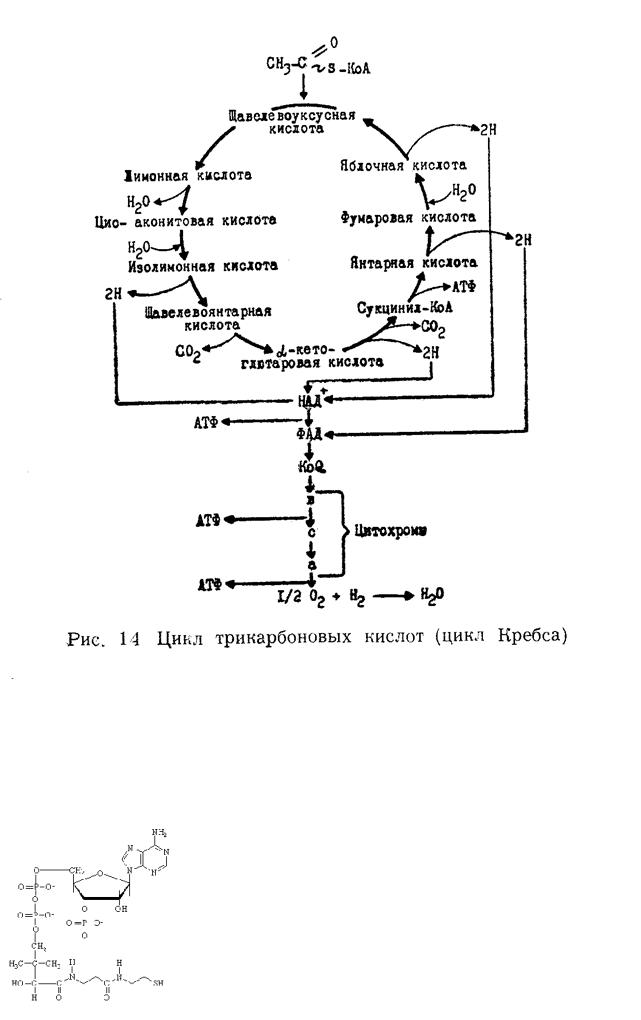
43.Строение коэнзима А, участие в обмене веществ.
HS-КоА входит в мультиферментный комплекс окислительного декарбоксилирования ПВК. Участвует в образовании активных форм жирных кислот (бета-окисление).
Строение – пантотеновая кислота, тиоэтанол амин, АМФ, в третьем положении дополнительный остаток фосфорной кислоты. Очень важен АцКоА – это промежуточный продукт окисления белков, жиров, углеводов. Образуется при окислительном декарбоксилировании ПВК, он запускает ЦТК. Из а-кетоглутората в ЦТК в процессе окислительного декарбоксилирования образуется сукцинилКоА, который идет на синтез гема.
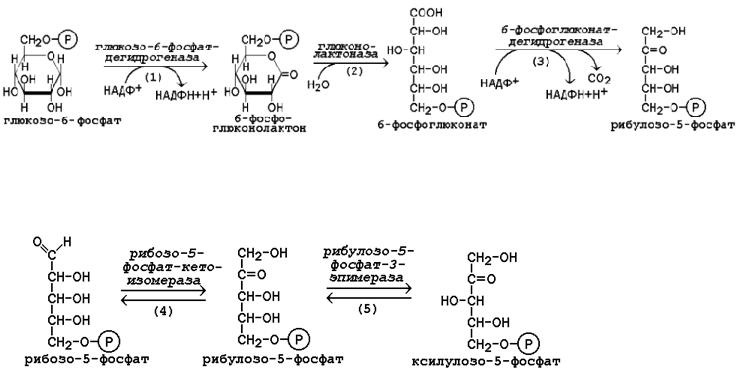
44. Пентозофосфатный путь окисления глюкозы.
Пентозофосфатный путь представляет собой прямое окисление глюкозы и протекает в цитоплазме клеток. Наибольшая активность ферментов пентозофосфатного пути обнаружена в клетках печени, жировой ткани, коры надпочечников, молочной железы в период лактации, зрелых эритроцитах. Низкий уровень этого процесса выявлен в скелетных и сердечной мышцах, мозге, щитовидной железе, легких.
Пентозофосфатный путь называют также апотомическим путѐм, так как в его реакциях происходит укорочение углеродной цепи гексозы на один атом, который включается в молекулу СО2.
Пентозофосфатный путь выполняет в организме две важнейшие метаболические функции:
он является главным источником НАДФН для синтеза жирных кислот, холестерола, стероидных гормонов, микросомального окисления; в эритроцитах НАДФН используется для восстановления глутатиона – вещества, препятствующего пероксидному гемолизу; он является главным источником пентоз для синтеза нуклеотидов, нуклеиновых кислот, коферментов (АТФ, НАД, НАДФ, КоА-SН и др.).
В пентозофосфатном пути можно выделить две фазы - окислительную и неокислительную.
Исходным субстратом окислительной фазы является глюкозо-6-фосфат, который непосредственно подвергается дегидрированию с участием НАДФ-зависимой дегидрогеназы. Продукт реакции гидролизуется , а образующийся 6- фосфоглюконат дегидрируется и декарбоксилируется . Таким образом, происходит укорочение углеродной цепи моносахарида на один углеродный атом («апотомия»), и образуется рибулозо-5-фосфат.
Неокислительная фаза пентозофосфатного пути начинается с реакций изомеризации. В ходе этих реакций одна часть рибулозо-5-фосфата изомеризуется в рибозо-5-фосфат, другая - в ксилулозо-5-фосфат.
Следуюшая реакция протекает при участии фермента транскетолазы, коферментом которой является тиаминдифосфат (производное витамина B1). В этой реакции происходит перенос двухуглеродного фрагмента с ксилулозо-5-фосфата на рибозо-5-фосфат:
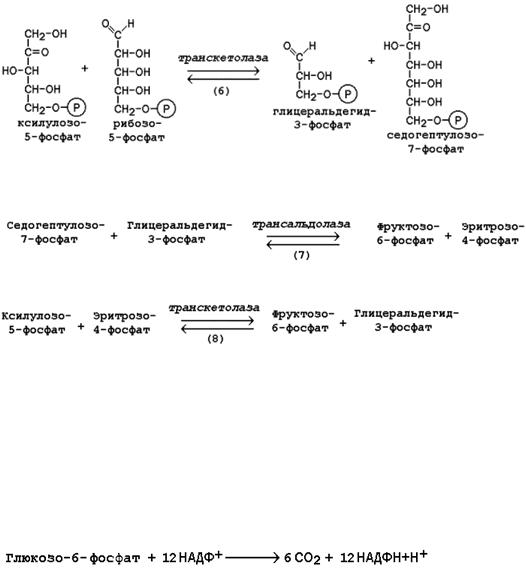
Образовавшиеся продукты взаимодействуют между собой в реакции, которая катализируется трансальдолазой и заключается в переносе остатка дигидроксиацетона на глицеральдегид-3-фосфат.
Продукт этой реакции эритрозо-4-фосфат участвует во второй транскетолазной реакции вместе со следующей молекулой ксилулозо-5-фосфата:
Таким образом, три молекулы пентозофосфатов в результате реакций неокислительной стадии превращаются в две молекулы фруктозо-6-фосфата и одну молекулу глицеральдегид-3-фосфата. Фруктозо-6-фосфат может изомеризоваться в глюкозо-6-фосфат, а глицеральдегид-3-фосфат может подвергаться окислению в гликолизе или изомеризоваться в дигидроксиацетонфосфат. Последний вместе с другой молекулой глицеральдегид-3-фосфата может образовывать фруктозо-1,6-дифосфат, который также способен переходить в глюкозо-6-фосфат.
Посредством пентозофосфатного пути может происходить полное окисление глюкозо-6-фосфата до шести молекул СО2. Все эти молекулы образуются из С-1-атомов шести молекул глюкозо-6-фосфата, а из образовавшихся при этом шести молекул рибулозо-5-фосфата снова регенерируются пять молекул глюкозо-6-фосфата:
Таким образом, полное окисление 1 молекулы глюкозы в пентозофосфатном пути сопровождается восстановлением 12 молекул НАДФ.
45. Роль печени в обмене углеводов. В печени происходит синтез гликогена, глюконеогенез, ПФЦ. Печеночный гликоген участвует в поддержании уровня глюкозы в крови, а гликоген мышц играет энергетическую роль. Глюкоза6фосфат идет на синтез гликогена.
46. Биосинтез глюкозы – глюконеогенез.
Глюконеогенез - биосинтез глюкозы из различных соединений неуглеводной природы. Биологическая роль глюконеогенеза заключается в поддержании постоянного уровня глюкозы в крови, что необходимо для нормального энергообеспечения тканей, для которых характерна непрерывная потребность в углеводах. Особенно это касается центральной нервной системы.
Роль глюконеогенеза возрастает при недостаточном поступлении углеводов с пищей. Так, в организме голодающего человека может синтезироваться до 200 г глюкозы в сутки. Глюконеогенез быстрее, чем другие метаболические процессы, реагирует на изменения диеты: введение с пищей большого количества белков и жиров активизирует процессы глюконеогенеза; избыток углеводов, наоборот, тормозит новообразование глюкозы.
Интенсивные физические нагрузки сопровождаются быстрым истощением запасов глюкозы в организме. В этом случае глюконеогенез является основным путѐм пополнения углеводных ресурсов, предупреждая развитие гипогликемии. Глюконеогенез в организме тесно связан также с процессами обезвреживания аммиака и поддержанием кислотноосновного баланса.
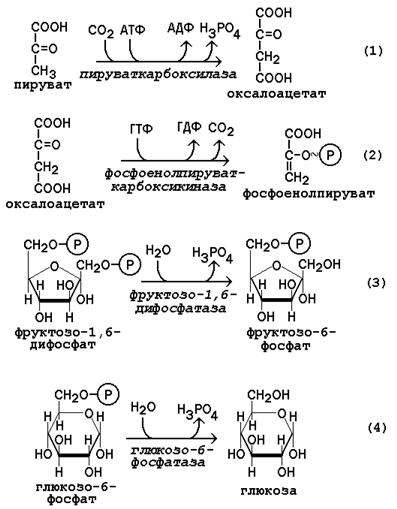
Последовательность реакций глюконеогенеза представляет собой обращение соответствующих реакций гликолиза. Лишь
три реакции гликолиза необратимы вследствие происходящих в ходе их значительных энергетических сдвигов: а) фосфорилирование глюкозы; б) фосфорилирование фруктозо-6-фосфата;
в) превращение фосфоенолпирувата в пируват.
Обход этих энергетических барьеров обеспечивают ключевые ферменты глюконеогенеза.
Обратный переход пирувата в фосфоенолпируват требует участия двух ферментов. Первый из них – пируваткарбоксилаза - катализирует реакцию образования оксалоацетата (рисунок 16.4, реакция 1). Коферментом пируваткарбоксилазы является биотин (витамин Н). Реакция протекает в митохондриях. Роль еѐ заключается также в пополнении фонда оксалоацетата для цикла Кребса.
Все последующие реакции глюконеогенеза протекают в цитоплазме. Мембрана митохондрий непроницаема для оксалоацетата, и он переносится в цитоплазму в виде других метаболитов: малата или аспартата. В цитоплазме указанные соединения вновь переходят в оксалоацетат. При участии фосфоенолпируваткарбоксикиназы из оксалоацетата образуется фосфоенолпируват (рисунок 16.4, реакция 2).
Фосфоенолпируват в результате обращения ряда реакций гликолиза переходит во фруктозо-1,6-дифосфат. Превращение фруктозо-1,6-дифосфата во фруктозо-6-фосфат катализируетсяфруктозодифосфатазой (рисунок 16.4, реакция 3). Фруктозо-6-фосфат изомеризуется в глюкозо-6-фосфат. Заключительной реакцией глюконеогенеза является гидролиз глюкозо-6-фосфата при участии фермента глюкозо-6-фосфатазы (рисунок 16.4, реакция 4).
Основными источниками глюкозы в глюконеогенезе являются лактат, аминокислоты, глицерол и метаболиты цикла Кребса.
Лактат – конечный продукт анаэробного окисления глюкозы. Может включаться в глюконеогенез после окисления до пирувата в лактатдегидрогеназной реакции.При продолжительной физической работе основным источником лактата является скелетная мускулатура, в клетках которой преобладают анаэробные процессы. Накопление молочной кислоты в мышцах ограничивает их работоспособность. Это связано с тем, что при повышении концентрации молочной кислоты в ткани снижается уровень рН (молочнокислый ацидоз). Изменение рН приводит к ингибированию ферментов важнейших
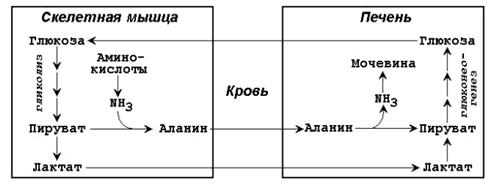
метаболических путей. В утилизации образующейся молочной кислоты важное место принадлежитглюкозо-лактатному циклу Кори (рисунок 16.5).
Глюкогенные аминокислоты, к которым относятся большинство белковых аминокислот. Ведущее место в глюконеогенезе среди аминокислот принадлежит аланину, который может превращаться в пируват путѐм трансаминирования. При голодании, физической работе и других состояниях в организме функционирует глюкозоаланиновый цикл, подобный циклу Кори для лактата (рисунок 16.2). Существование цикла аланин – глюкоза препятствует отравлению организма, так как в мышцах нет ферментов, утилизирующих аммиак. В результате тренировки мощность этого цикла значительно возрастает.
Другие аминокислоты могут, подобно аланину, превращаться в пируват, а также в промежуточные продукты цикла Кребса (α-кетоглутарат, фумарат, сукцинил-КоА). Все эти метаболиты способны преобразовываться в оксалоацетат и включаться в глюконеогенез.
47. Регуляция концентрации глюкозы в крови, пути поступления и расходования глюкозы, гипо- и гипергликемия.
Уровень глюкозы в крови регулирует ряд гормонов. Инсулин понижает уровень сахара в крови, а все остальные повышают его – адреналин, глюкогон, глюкокортикоиды, соматотропный гормон. Глюкоза в организм человека поступает с пищей и образуется в результате протекания следующих процессов: распад гликогена, глюконеогенез. Глюкоза расходуется на: 1) синтез гликогена 2) ПФЦ (рибоза5фосфат + НАДФН) 3) гликолиз, в результате образуется 2 ПВК (высвобождается 8 АТФ). Далее ПВК в анаэробных условиях превращается в 2 МК и 2 АТФ. В аэробны условиях происходит окислительное декарбоксилирование ПВК до 2 АцКоА (6 АТФ), который запускает ЦТК, в результате чего образуется СО2, Н2О и 24 АТФ. Кортизол угнетает синтез белков в тканях и не использованные на синтез а/к идут на глюконеогенез, а в печени усиливается биосинтез ключевого фермента глюконеогенеза пируваткарбоксилазы. Возрастные показатели уровня сахара в крови. Норма взрослого человека – 3,5-5,5 ммоль/л, если больше 11 ммоль/л, то сахар появляется в моче – это пороговое вещество. Если меньше 3,3 ммоль/л – гипогликемия, больше 6 ммоль/л – гипергликемия. Гипергликемия – причины: физиологическая гипергликемия – алиментарная, эмоциональная; при сахарном диабете; при гипертиреозе, адренокортицизме, гиперпитуитаризме. Гипогликемия – причины: длительное голодание; нарушение всасывания (заболевания ЖКТ), хронические заболевания печени (нарушение синтеза гликогена); нарушение секреции контринсулярных гормонов – гипопитуитаризм, хроническая недостаточность коры надпочечников; гипотиреоз; заболевания ЦНС (инсульты); передозировка инсулина и пероральных диабетических средств; нарушение режима питания у больных с сахарным диабетом; заболевания поджелудочной железы (инсулинома).
Сахарный диабет. При недостаточности содержания инсулина возникает сахарный диабет: повышается концентрация глюкозы в крови, появляется глюкоза в моче и уменьшается содержание гликогена в печени. При введении инсулина больным диабетом происходит коррекция метаболических сдвигов. Инсулин контролирует эти процессы на генетическом уровне как индуктор синтеза ключевых ферментов гликолиза: гексокиназы, фосфофруктокиназы, пируваткиназы. Инсулин также индуцирует синтез гликогенсинтетазы. Одновременно инсулин действует как репрессор синтеза ключевых ферментов глюконеогенеза. Инсулин синтезируется в
поджелудочной железе в виде препроинсулина, состоит из альфа цепи – 20 а/к и бета цепи – 31 а/к. Рецепторы инсулина имеют сложное строение. Состоят из 2х альфа субъединиц, которые не проникают через клеточную мембрану и 2х бета субъединиц, которые находятся в цитозоле. При сахарном диабете может нарушаться передача трансмембранного сигнала, и в этом случае инсулин не помогает. Повышение сахара в крови в течении 2-3х минут вызывает секрецию инсулина, 40мин-1час пока не станет содержание сахара в крои в норме. Причины развития сахарного диабета с биохимической позиции: 1) повышение активности тканевого фермента инсулиназы - происходит восстановление или окисление дисульфидных мостиков, инсулин теряет свою активность 2) замена ряда а/к в полипептидных цепях инсулина; если заменить фенилаланин на лейцин с С-конца бета цепи, происходит потеря гормональной активности в 10 раз 3) дефект рецепторов инсулина – нарушается связь инсулина с рецепторами или связь происходит, но передача с альфа на бета субъединицы не происходит 4) нарушено превращение проинсулина в инсулин – нарушение отщепления С пептида. Биохимические нарушения при сахарном диабете: первый симптом - манифестный симптом – нарушается транспорт глюкозы через клеточную мембрану, глюкоза не транспортируется из крови в клетки, это приводит к превышению сахара в крови – гипергликемия. Мало инсулина – это приводит к повышению синтеза контринсулярных гормонов и повышение синтеза кортизола – усиливаются процессы глюконеогенеза – наблюдается много сахара в крови. Норма сахара в крови – 3,3-5,5 ммоль/л, порог почечной проницаемости для глюкозы – 11 ммоль/л, при превышении порога сахар обнаруживается в моче – глюкозурия, это приводит к повышению осмоляльности, наблюдается приток жидкости из тканей – полиурия, появляется полидипсия – глубокое нарушение. Полиурия
– с мочой выводится большое количество ионов Na, K, Ca, фосфатов, что вызывает увеличение концентрации контринсулярных гормонов. В результате этого происходит потеря ионов – изменяется буферная емкость крови, снижается концентрация 2,3дифосфоглицератов, это приводит к нарушению отдачи кислорода к тканям (гипоксия тканей).
48.Нарушение обмена моно- и дисахаридов.
Фруктоземия
Заболевание обусловлено врожденным отсутствием ферментов фруктозофосфатальдолазы и фруктозодифосфатальдолазы. Избыточное накопление фруктозофосфата нарушает гликогенолиз, что приводит к гипогликемии. В печени имеется недостаточное количество фермента фруктозо-1- фосфат-альдолазы, в результате продукты обмена (фруктозо-1-фосфат) накапливаются в организме (печени, почках, слизистых оболочках кишечника) и оказывают повреждающее действие. Морфологически в печени выявляются жировая инфильтрация, умеренный перилобулярный фиброз.
Клиническая картина. Симптомы возникают при введении в рацион сладкой пищи или фруктовых соков, т.е. продуктов, содержащих фруктозу. Со 2–4-го месяца развиваются диспептические явления и состояния острой гипогликемии, которые проявляются бледностью, вялостью, потливостью, запахом ацетона. В тяжелых случаях может развиться гипогликемическая кома с потерей сознания и судорогами. Характерно, что гипогликемия возникает после приема пищи. С возрастом дети сами отказываются от сладкой пищи. Постоянным признаком является гепатомегалия (обычно с увеличением обеих долей) с ровным краем и некоторым уплотнением печени.
Лечение. Диета, лишенная фруктозы, главными источниками которой считаются мед, сахарный тростник и свекла, фрукты, джемы, повидло, морковь, какао, цикорий, репа.
Новорожденным назначают молочные смеси без сахара.
Дети первого года получают молочные смеси, содержащие только лактозу и декстрин-мальтозу.
Галактоземия
Наследственная энзимопатия. Наследуется по рецессивному типу. В основе галактоземии лежит нарушение обмена галактозы в связи с отсутствием фермента галактозофосфат-
уридилтрансферазы. В результате в крови накапливаются в больших концентрациях галактоза и галактозофосфат. Происходит нарушение процесса ферментативного превращения галактозы в глюкозу с накоплением галактозы и продуктов ее обмена в клетках, что оказывает повреждающее действие на функции печени, головного мозга, хрусталика глаза, почек.
Клинические признаки заболевания возникают рано — через 1–2 недели после рождения ребенка. Пропадает аппетит, появляются вялость, рвота, понос. Наблюдается дефицит массы тела. Постепенно развивается гепато-, спленомегалия, появляется стойкая гипербилирубинемия, преимущест-венно за счет прямого билирубина. Часто отмечается катаракта, ведущая к слепоте. Могут быть симптомы, свидетельствующие о поражении почек (протеинурия, гипераминоацидурия), центральной нервной системы (задержка психофизического развития). После чайно-водной паузы состояние улучшается, но введение молока обусловливает рецидив нарушений со стороны желудочно-кишечного тракта. При несвоевременной диагностике заболевание прогрессирует, что приводит к тяжелым последствиям или летальному исходу.
Лечение. Диетотерапия является единственным методом лечения. Для вскармливания ребенка используют смеси, лишенные лактозы. Из питания детей более старшего возраста исключают цельномолочные продукты.
Гликогенозы
Группа наследственных болезней обмена полисахаридов, развивающихся в результате нарушения синтеза или распада гликогена на простые сахара. При этом нормальный и аномальный гликоген одновременно накапливаются в клетках печени и других органах и тканях.
Клиническая картина гликогенозов характеризуется гипогликемией (рвота, судороги, потеря сознания, кома). Течение болезни зависит от места депонирования гликогена: печень, почки, мышечная ткань. Соответственно выделяют цирроз печени, почечную форму, мышечный синдром (включая сердечную форму). Преобладание у новорожденного ребенка симптомов гипогликемии может привести к синдрому внезапной смерти. Прогноз зависит от типа болезни.
Гликогеноз 0 типа (агликеноз) характеризуется резким снижением запасов гликогена в печени, наблюдается тяжелое состояние вплоть до развития комы (гипогликемический синдром). Кома может возникать после рождения при позднем прикладывании ребенка к материнской груди, а позднее — утром натощак и в перерывах между кормлениями. При отсутствии лечения ребенка наступает нарушение психомоторного развития.
Гликогеноз I типа (болезнь Гирке) — гликогеноз, обусловленный недостаточностью глюкозо-6- фосфатазы, приводящей к невозможности превращения глюкозо-6-фосфата в глюкозу, что сопровождается накоплением гликогена в печени и почках; наследуется по аутосомнорецессивному типу. Дефект фермента в печени, почках, слизистой оболочке тонкой кишки. При его первых проявлениях наблюдаются отсутствие аппетита, рвота, респираторный дистресссиндром, гипогликемические судороги (кома), которые выявляются сразу после рождения или в грудном возрасте.
Гликогеноз II типа (болезнь Помпе) — наследуется по аутосомно-рецессивному типу. Симптомы проявляются в первые недели жизни — до шести месяцев после рождения. Дефект фермента найден в печени, почках, селезенке, мышцах, нервной ткани, лейкоцитах. Наблюдается расстройство дыхания, беспокойство или адинамия. Отмечаются отсутствие аппетита, задержка роста, мышечная гипотония. Увеличиваются размеры сердца, печени, почек, селезенки. Сердце приобретает шаровидную форму, в связи с гипертрофией миокарда появляются изменения ЭКГ. Часто возникают гипостатические пневмонии, бронхиты, ателектазы легких, наблюдаются миодистрофия, гипорефлексия, спастические параличи. Мышечная форма гликогеноза II типа возникает только в мышцах при дефиците кислой α-1,4-глюкозидазы. Болезнь проявляется в более поздние сроки и по клинической картине напоминает миопатию.
Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз) — гликогеноз, вызванный недостаточностью фермента амило-1,6-глюкозидазы. Этот фермент катализирует расщепление связей […C-O-C…] в молекуле гликогена в точках ветвления. Болезнь сопровождается отложением атипичного гликогена в печени, сердце, мышцах. Наследуется по аутосомно-рецессивному типу.
Дефект фермента найден в печени, мышцах, лейкоцитах, эритроцитах. С первых месяцев жизни ребенка наблюдаются гепатомегалия, мышечная гипотония, гипертрофия отдельных групп мышц. В некоторых случаях у больных отмечаются нарушение сердечной проводимости и кровообращения, гипертрофия миокарда. Развитие заболевания замедляется после пятилетнего возраста или в пубертатном периоде.
Гликогеноз IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени) — гликогеноз, семейный цирроз печени, вызванный дефектом фермента амило-(1,4-1,6)- трансглюкозидазы. Этот фермент катализирует превращение 1,4-связей в молекуле гликогена в 1,6-связи, то есть обусловливает ветвление молекулы полисахарида. Заболевание сопровождается избыточным накоплением атипичного гликогена в печени. Наследуется по аутосомно-рецессивному или связанному с полом типу. Дефект фермента найден в печени, почках, мышцах, лейкоцитах. Болезнь наблюдается с первых месяцев жизни и характеризуется гепатоспленомегалией, развитием цирроза печени, желтухой, гипогликемией.
Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность) — гликогеноз, связанный с дефектом мышечной фосфорилазы. Заболевание, обусловленное нарушением каталитической функции этого фермента; сопровождается отложением значительного количества гликогена в мышцах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в мышцах. В связи с гликогенной инфильтрацией скелетные мышцы увеличиваются в объеме, становятся очень плотными. Мышечная слабость, мышечные спазмы, тахикардия при физической нагрузке появляются в первые десять лет жизни и прогрессируют. Наблюдается транзиторная миоглобинурия. Концентрация лактата в крови после физической нагрузки уменьшается. Чаще (в 5 раз) болеют лица мужского пола.
Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность) — гликогеноз, вызванный недостаточностью фосфорилазы печени. Фосфорилаза печени катализирует фосфорилирование гликогена с образованием глюкозо-1-фосфата. Нарушение этого механизма приводит к избыточному отложению гликогена в печени. Наследуется, предположительно, по аутосомно-рецессивному типу. Проявляется обычно на первом году жизни. Характерны значительное увеличение печени в результате гликогенной инфильтрации гепатоцитов, задержка роста, кукольное лицо, гиперлипидемия, гипергликемия после внутривенного введения галактозы, повышенное содержание гликогена в эритроцитах. Наследуется по аутосомнорецессивному типу. Дефект фермента найден в печени, лейкоцитах. Проявляется обычно на первом году жизни.
Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность) — симптомы сходны с гликогенозом V типа. Дефект фермента найден в мышцах, эритроцитах. Также характерны мышечная слабость, утомляемость и отсутствие гиперлактацидемии после физической нагрузки.
Гликогеноз VIII типа (болезнь Томсона) — встречается редко, наследование не установлено. Дефект фермента найден в печени, в головном мозге. После рождения увеличиваются размеры печени, появляются нистагм («танцующие глаза») и атаксия. Неврологическая симптоматика прогрессирует.
Гликогеноз IX типа (болезнь Хага) — наследуется по рецессивному, связанному с полом типу. Дефект фермента найден в печени. У больных наблюдается гепатомегалия.
Гликогеноз Х типа — известен случай у единственного больного, наследование не установлено. Дефект фермента найден в печени, мышцах. Наблюдалась гепатомегалия, через 6 лет после начала заболевания появились мышечные боли и спазмы мышц после физических упражнений.
Гликогеноз XI типа (болезнь Фанкони — Бикеля) — наследование не установлено. Дефект фермента найден в печени, почках. Характеризуется значительным увеличением печени и резкой задержкой роста. Наблюдаются симптомы гипофосфатемического рахита. В пубертатном периоде возможны уменьшение размеров печени, ускорение роста, нормализация уровня фосфора в крови.
Лечение гликогенозов в основном симптоматическое и направлено на изменение нарушений обменных процессов. Цель лечения — предупредить тяжелую гипогликемию. Назначают диету, богатую белками и углеводами. Питание должно быть частым (каждые 4 ч). Белки служат источником аминокислот — субстратов глюконеогенеза; они уменьшают углеводную нагрузку,
приводящую к гипергликемии и лактат-ацидозу. Такая диета предотвращает гипогликемию и кетоацидоз натощак, уменьшает гипергликемию и лактат-ацидоз после еды и способствует ускорению роста. При мышечных формах гликогенозов улучшение отмечается при соблюдении диеты с высоким содержанием белка, назначении фруктозы, поливитаминов, АТФ.
49. Современные данные об активных формах углеводов, жирных кислот и аминокислот.
Глюкоза 6 фосфат (учавствует в синтезе гликогена) УДФгалактоза(промежуточный продукт при метаболизме галактозы), УДФглюкоза(ключевого метаболита углеводного обмена ) - активные формы углеводов · Активация жирных кислот.
Свободная жирная кислота независимо от длины углеводородной цепи является метаболически инертной и не может подвергаться никаким биохимическим превращениям, в том числе окислению, пока не будет активирована. Активация жирной кислоты протекает на наружной поверхности мембраны митохондрий при участии АТФ, коэнзима A (HS-KoA) и ионов Mg2+. Реакция катализируется ферментом ацил-КоА-синтетазой:
В результате реакции образуется ацил-КоА, являющийся активной формой жирной кислоты.
Считают, что активация жирной кислоты протекает в 2 этапа. Сначала жирная кислота реагирует с АТФ с образованием ациладенилата, представляющим собой эфир жирной кислоты и АМФ. Далее сульфгидрильная группа КоА действует на прочно связанный с ферментом ациладенилат с образованием ацил-КоА и АМФ.
Активация жирных кислот и их проникновение из цитоплазмы в митохондрии. Образование "активной формы" жирной кислоты (ацил-КоА) из коэнзима А и жирной кислоты является эндергоническим порцессом протекающим за счет использования энергии АТФ:
Реакция катализируется ацил-КоА-синтетазой. Существует несколько таких ферментов: один из них катализирует активацию жирных кислот, содержащих от 2 до 3 углеродных атомов, другойот 4 до 12 атомов, третий - от 12 и более атомов углерода.
Как уже отмечалось, окисление жирных кислот (ацил-КоА) происходит в митохондриях. В последние годы было показано, что способность ацил-КоА проникать из цитоплазмы в митохондрии резко возрастает в присутствии азотистого основания - карнитина (γ-триметиламино-β-гидроксибутирата). АцилКоА, соединяясь с карнитином, при участии специфического цитоплазматического фермента (карнитин-ацил-КоА-трансферазы) образует ацилкарнитин (эфир карнитина и жирной кислоты), который обладает способностью проникать внутрь митохондрии:
После прохождения ацилкарнитина через мембрану митохондрии происходит обратная реакция - расщепление ацилкарнитина при участии HS-KoA и митохондриальной карнитин-ацил-КоА-трансферазы:
При этом карнитин возвращается в цитоплазму клетки, а ацил-КоА подвергается в митохондриях окислению.
· Активация аминокислот Для каждой из 20 аминокислот имеется соответствующая аминоацил-тРНК-лигаза,
которая в цитоплазме соединяет аминокислоту с тPHK(tRNA) (см. с. 88). Этот процесс активации аминокислот осуществляется в две стадии. Сначала аминокислота связывается с ферментом и реагирует с АТФ (АТР), образуя макроэргический смешанный ангидрид — аминоациладенилат. Затем аминоацильный остаток переносится на концевую 3'-ОН-группу концевого