

Документ Microsoft Word
.docxИнфракрасная спектроскопия
[править | править вики-текст]
Материал из Википедии — свободной энциклопедии

Инфракрасный спектрометр
Инфракрасная спектроскопия (колебательная спектроскопия, ИК-спектроскопия, средняя инфракрасная спектроскопия, ИКС) — раздел спектроскопии, изучающий взаимодействие инфракрасного излучения с веществами.
При
пропускании инфракрасного излучения
через вещество происходит
возбуждение колебательных
движений молекул или
их отдельных фрагментов ![]() .
При этом наблюдается ослабление
интенсивности света, прошедшего через
образец. Однако поглощение происходит
не во всём спектре падающего излучения,
а лишь при тех длинах
волн,
энергия которых соответствует энергиям
возбуждения колебаний в изучаемых
молекулах. Следовательно, длины волн
(или частоты), при которых наблюдается
максимальное поглощение ИК-излучения,
могут свидетельствовать о наличии в
молекулах образца тех или иных функциональных
групп и
других фрагментов
.
При этом наблюдается ослабление
интенсивности света, прошедшего через
образец. Однако поглощение происходит
не во всём спектре падающего излучения,
а лишь при тех длинах
волн,
энергия которых соответствует энергиям
возбуждения колебаний в изучаемых
молекулах. Следовательно, длины волн
(или частоты), при которых наблюдается
максимальное поглощение ИК-излучения,
могут свидетельствовать о наличии в
молекулах образца тех или иных функциональных
групп и
других фрагментов ![]() ,
что широко используется в различных
областях химии для установления структуры
соединений.
,
что широко используется в различных
областях химии для установления структуры
соединений.
Экспериментальным результатом в ИК-спектроскопии является инфракрасный спектр — функция интенсивности пропущенного инфракрасного излучения от его частоты. Обычно инфракрасный спектр содержит ряд полос поглощения, по положению и относительной интенсивности которых делается вывод о строении изучаемого образца. Такой подход стал возможен благодаря большому количеству накопленной экспериментальной информации: существуют специальные таблицы, связывающие частоты поглощения с наличием в образце определённых молекулярных фрагментов. Созданы также базы ИК-спектров некоторых классов соединений, которые позволяют автоматически сравнивать спектр неизвестного анализируемого вещества с уже известными и таким образом идентифицировать это вещество.
Инфракрасная
спектроскопия является ценным
аналитическим методом и служит для
исследования строения органических
молекул ![]() , неорганических икоординационных
, неорганических икоординационных ![]() ,
а также высокомолекулярных
соединений
,
а также высокомолекулярных
соединений ![]() .
Основным прибором, используемым для
подобных анализов, является инфракрасный
спектрометр(дисперсионный
.
Основным прибором, используемым для
подобных анализов, является инфракрасный
спектрометр(дисперсионный ![]() или
с преобразованием
Фурье
или
с преобразованием
Фурье ![]() ).
).
Анализ
сложных образцов стал возможен благодаря
разработке новых техник инфракрасной
спектроскопии: ИК-спектроскопии
отражения ![]() и
ИК-микроскопии
и
ИК-микроскопии ![]() .
Кроме того инфракрасная спектроскопия
была объединена с другими аналитическими
методами: газовой
хроматографией и термогравиметрией
.
Кроме того инфракрасная спектроскопия
была объединена с другими аналитическими
методами: газовой
хроматографией и термогравиметрией ![]() .
.
Содержание
[убрать]
-
1 История метода
-
2 Принцип метода
-
2.1 Основные характеристики ИК-излучения
-
2.2 Виды и энергия колебаний молекул
-
2.3 Характеристические колебания
-
2.4 Поглощение излучения
-
-
3 ИК-спектрометры
-
3.1 Дисперсионные ИК-спектрометры
-
3.2 Спектрометры с преобразованием Фурье
-
-
4 ИК-спектроскопия пропускания
-
4.1 Органические соединения
-
4.2 Неорганические, координационные и металлоорганические соединения
-
4.3 Высокомолекулярные соединения
-
4.4 Подготовка образцов
-
-
5 ИК-спектроскопия отражения
-
5.1 Спектроскопия НПВО
-
5.2 Спектроскопия внешнего отражения
-
-
6 ИК-микроскопия
-
7 Комбинирование с другими методами
-
7.1 С газовой хроматографией
-
7.2 С термогравиметрическим анализом
-
-
8 Применение
-
8.1 Исследование памятников искусства
-
8.2 Применение в медицине
-
8.3 Применение в судебной экспертизе
-
-
9 См. также
-
10 Примечания
-
11 Литература
-
12 Ссылки
-
12.1 Базы данных ИК-спектров
-
12.2 Учебные материалы
-
История метода[править | править вики-текст]

Уильям Кобленц
Инфракрасное излучение было открыто в 1800 году астрономом Уильямом Гершелем. Используя призму, он наблюдал повышение температуры в области, находящейся за красной границей спектра видимого излучения. В 1882—1900 годах Уильям Эбни и Эдвард Фестинг[en] записали инфракрасные спектры 52 соединений и сопоставили наблюдаемые полосы поглощения с функциональными группами, присутствующими в этих молекулах. Cущественный вклад в метод сделал американский физик Уильям Кобленц, который с 1903 года, пользуясь призмой из хлорида натрия, получил весьма точные и полные ИК-спектры для сотен органических и неорганических веществ[1][2].
Первые эксперименты по регистрации инфракрасных спектров были крайне трудоёмкими, поскольку исследователи были вынуждены собирать собственные приборы, шлифовать и полировать призмы, серебрить зеркала, градуировать приборы по показателям преломлениякаменной соли. При этом спектрометры были чувствительны к вибрациям, поэтому их располагали на фундаменте, а исследования проводили по ночам. Время регистрации одного спектра составляло от 3 до 4 часов. Уже в ранних работах было показано, что ИК-спектры соединений имеют индивидуальный вид[1].
В то время природа поглощения инфракрасного излучения не была до конца ясна, однако к 1930-м годам была создана теория, в которой полагалось, что это поглощение наблюдается вследствие колебаний молекул и что характер этого поглощения так или иначе связан с изменением дипольного момента, правилами отбора, симметрией молекул и степенью ангармонизма колебаний[2].
В 1940 году фирмы Dow Chemical и American Cyanamid создали собственные однолучевые приборы для изучения углеводородов. Коммерческие спектрометры стали выпускаться в 1946 году при сотрудничестве American Cyanamid с Perkin-Elmer[en]. Доступность приборов привела к созданию обширных таблиц корреляции наблюдаемых полос поглощения со структурой поглощающих функциональных групп[3].
После Второй мировой войны появилась возможность усиливать слабый сигнал ИК-спектрометров, что сократило время эксперимента до 1—2 часов. Затем была усовершенствована техника изготовления термоэлектрических приёмников с малым временем отклика. Эти улучшенные детекторы позволяли избежать дрейфа показаний во времени и привели к созданию двухлучевых приборов, где шкала калибровалась в процентах пропускания против шкалы длин волн или волновых чисел[1].
Стало возможным промышленное получение больших и качественных кристаллов галогенидов щелочных металлов, необходимых для создания оптических элементов приборов, что позволило преодолеть многие трудности. Например, синтетический бромид калия, в отличие от использовавшейся ранее каменной соли, позволил записывать ИК-спектры вплоть до 400 см–1, тогда как прежний предел составлял 650 см–1[4].
Расцветом ИК-спектроскопии стало появление ИК-интерферометров, которые первоначально использовались для детектирования очень слабого инфракрасного излучения астрономических объектов. После разработки быстрых методов конвертирования интерферограмм в спектры (преобразование Фурье) и уменьшения времени сканирования подобные приборы стали выпускаться серийно, что в 1970-е годы позволило выйти на рынок ИК-спектрометров компаниям, которые производили компьютеры, но не имели опыта работы в области спектроскопии (Nicolet, Bruker). Преимущество ИК-интерферометров заключалось в их мультиплексности (преимущество Фелгетта), то есть одновременном сборе информации о поглощении всех длин волн, за счёт чего достигалось более высокоеотношение сигнала к шуму для фиксированного времени сканирования спектра. Второе преимущество состояло в производительности приборов нового типа: в то время как дисперсионные приборы имели вход и выход, которые ограничивали количество проходящего через них света, производительность интерферометра определялась толщиной пучка света от источника. Вероятно, мода также сыграла значительную роль в распространении ИК-спектрометров с преобразованием Фурье, поскольку в то время не существовало большой необходимости в высоком соотношении сигнал/шум: образцы обычно готовились гораздо дольше, чем проводилось измерение, и масса образцов была достаточной для записи качественных спектров[5].
ИК-интерферометры позволили получать спектры в дальней ИК-области, наблюдать решёточные колебания кристаллов, а также, благодаря высокому отношению сигнал/шум, преодолевать сложности при интерпретации спектров органических соединений. Одним из популярных занятий в то время была цифровая обработка спектров, а именно удаление полос поглощения растворителей, определение степени чистоты и характера примесей. Интерферометры нашли широкое применение в исследовании водных растворов биологических молекул[6].
В 1980-е годы появились комбинированные методы, объединившие газовую хроматографию и ИК-спектроскопию. Напольные приборы большого размера сменились более компактными настольными моделями. Появилась возможность ступенчатого сканирования во времени, что позволило изучать динамические процессы со сбором данных в одной точке[6].
Принцип метода[править | править вики-текст]
Основные характеристики ИК-излучения[править | править вики-текст]
Поглощение электромагнитного излучения
ИК-спектроскопия
основана на явлении поглощения химическими
веществами инфракрасного излучения с
одновременным возбуждением колебаний
молекул. Инфракрасное излучение
представляет собой электромагнитную
волну и
характеризуетсядлиной
волны λ, частотой ν
и волновым числом ![]() ,
которые связаны следующей зависимостью:
,
которые связаны следующей зависимостью:
![]()
где с — скорость света, а n — показатель преломления среды[7].
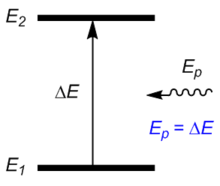
В спектроскопии поглощения, частным случаем которой является ИК-спектроскопия, происходит поглощение молекуламифотонов определённой энергии, которая связана с частотой электромагнитной волны через постоянную Планка:
![]()
При поглощении фотона происходит возбуждение — увеличение энергии молекулы: она переходит из основного колебательного состояния E1 в некоторое возбуждённое колебательное состояние E2 так, что энергетическая разница между этими уровнями равна энергии фотона[7].
![]()
Энергия поглощённого инфракрасного излучения расходуется на возбуждение колебательных переходов для веществ в конденсированном состоянии. Для газов поглощение кванта ИК-излучения приводит к колебательным и вращательным переходам[7].
Виды и энергия колебаний молекул[править | править вики-текст]
См. также: Молекулярные колебания
Колебательные движения молекул определяются их внутренними, или колебательными, степенями свободы. Число колебательных степеней свободы и соответствующих им нормальных[K 1] колебаний равно (3n–5) для линейных молекул и (3n–6) для нелинейных молекул, где n — число атомов в молекуле[K 2]. Например, молекула воды H2O нелинейна и имеет 3 колебательные степени свободы, а линейная молекула водорода H2 — лишь одну[8][9].
Колебания молекул могут заключаться в изменении длин связей (валентные колебания, v) либо углов между связям (деформационные колебания, δ). Валентные колебания могут быть симметричными и антисимметричными, а деформационные колебания подразделяются на ножничные, маятниковые, веерные икрутильные. Для более сложных молекул, в которых одна из деформационно колеблющихся частей гораздо массивнее другой, деформационные колебания чаще описывают как плоскостные и внеплоскостные. Колебания, которые заключаются в одновременном изменении нескольких длин связей или валентных углов, называются скелетными[10].
|
Валентные колебания |
Деформационные колебания |
||||
|
симметричное |
антисимметричное |
плоскостные |
внеплоскостные |
||
|
ножничное |
маятниковое |
веерное |
крутильное |
||
|
|
|
|
|
|
|




Потенциальные кривые для гармонического и ангармонического осцилляторов
Колебания молекул могут быть описаны при помощи моделей гармонического и ангармонического осциллятора. С точки зрения модели гармонического осциллятора, двухатомная молекула представляет собой две массы m1 и m2, соединённые упругой пружиной, не имеющей массы, с силовой постоянной K. В таком случае частота колебания атомов такой молекулы вдоль линии, проходящей через центры их масс, равна[11]:
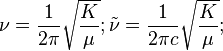
![]()
Из данных выражений следует, что наблюдаемая частота колебаний двухатомного осциллятора зависит от силовой постоянной K, которая, в свою очередь, связана с энергией связи между двумя атомами, а также от массы атомов, участвующих в колебании. Для многоатомных молекул колебания носят более сложный характер и приближение гармонического осциллятора неприменимо[11].
Потенциальная энергия гармонического осциллятора связана с отклонением расстояния между атомами X следующим образом[11]:
![]()
График потенциальной энергии представляет собой параболу, симметричную относительно начального положения атомов в состоянии покоя (re). Согласно квантовой механике, энергетические состояния молекулы квантованы, то есть являются дискретными. Подобные квантованные состояния называют колебательными уровнями. Колебательные уровни отстоят друг от друга на одинаковом расстоянии, и их энергия может быть вычислена по уравнению[11]
![]()
При vi = 0 молекула находится на самом низком колебательном уровне, и колебательная энергия в таком состоянии равна E = ½ hν. Данная энергия всегда присуща молекуле и не может быть отобрана. В приближении гармонического осциллятора разрешены лишь переходы с Δv = ±1, то есть лишь на соседние уровни (правило отбора)[11].
Более точной является модель ангармонического осциллятора. Ангармоничность проявляется, если величина дипольного момента изменяется не пропорционально смещению атомов. Отличие данной модели состоит в том, что расстояние между колебательными уровнями уменьшается с увеличением номера уровня. Отклонение от гармоничности также увеличивается снизу вверх. Энергия уровня в случае ангармонического осциллятора выражается следующим образом[11]:
![]()
Ангармоничность колебаний приводит к уменьшению строгости правила отбора, вследствие чего в спектрах могут наблюдаться переходы с Δv = ±2 — обертоны. Как правило, частота обертона попадает в область 2×ν1-b, где b = 2—10 см–1. Возможно также возникновение комбинационных, или составных, полос, имеющих частоту ν1+ ν2, где ν1 и ν2 — частоты каких-либо фундаментальных колебаний молекулы. Комбинационная полоса появляется при колебательных переходах из возбуждённых состояний. Обычно для конденсированного состояния интенсивность обертонов и комбинационных полос в 10—100 раз ниже, чем основных, хотя могут встречаться исключения[12].
Если обертон или комбинационная полоса совпадают по частоте с каким-либо фундаментальным колебанием, проявляется резонанс Ферми[en], который приводит к появлению двух полос поглощения примерно одинаковой интенсивности, в то время как ожидается наличие лишь одной фундаментальной полосы. Иногда также происходит смешивание колебаний с примерно одинаковой частотой: при этом число колебаний остаётся таким же, но они проявляются при других частотах и уже не могут быть отнесены только к одной связи. Осложняющим фактором также является появление в спектрах тонкой структуры, соответствующей вращательным переходам (такое явление наблюдается лишь для веществ в газообразном состоянии)[10].
Характеристические колебания[править | править вики-текст]
См. также: Таблица характеристических частот в инфракрасной спектроскопии
Многоатомные молекулы имеют 3n–6(5) нормальных колебаний, и в каждом таком колебании участвуют не пары атомов при одной связи, а в той или иной степени всеn атомов молекулы. Однако было экспериментально установлено, что для колебаний некоторых функциональных групп вклад «посторонних» атомов и связей достаточно мал, поэтому вне зависимости от окружения эти функциональные группы поглощают в ограниченном интервале частот. Этот факт позволил путём сравнения многочисленных спектров соотнести наличие в молекуле характерных фрагментов с наблюдаемыми полосами поглощения. Такие полосы получили название групповых, или характеристических. По ним можно быстро и однозначно подтвердить присутствие или отсутствие в молекуле соответствующих фрагментов[13].
Возникновение характеристических колебаний может происходить по двум причинам[14]:
-
Если характеристическое колебание относится к лёгкому атому, связанному с тяжёлым, то практически всё движение сосредоточено именно на нём, и влияние остальной части молекулы на него весьма слабое.
-
Колебания, относящиеся к атомам очень близкой массы (например, C=O, C≡N), слабо взаимодействуют с колебаниями остальных частей молекулы.
Существуют также менее определённые характеристические колебания, которые наблюдаются в сравнительно более широком интервале частот. Однако их положение в спектре можно объяснить массой атомов, резонансом или электронными эффектами в молекуле[14].
Поглощение излучения[править | править вики-текст]
Обычно в эксперименте прибор испускает одновременно все длины волн инфракрасного излучения, включая ближнюю ИК-область (14 000 — 4000 см–1), среднюю ИК-область (4000 — 400 см–1) и дальнюю ИК-область (400 — 10 см–1). Поглощение излучения веществом количественно описывается законом Бугера — Ламберта — Бера, а спектр получается при построении зависимости пропускания (T, англ. transmittance, %) или оптической плотности (D, англ. optical density) от длины волны (частоты, волнового числа)[15].
Для того, чтобы поглощение излучения произошло, необходимо выполнение двух условий. Во-первых, поглощаются лишь волны такой частоты, которая совпадает с частотой того или иного колебания молекулы. Во-вторых, колебание должно вызывать изменение дипольного момента молекулы. По этой причине молекулы, не имеющие дипольного момента (например, H2, N2, O2, а также соли без ковалентных связей и металлы), не поглощают инфракрасное излучение. Интенсивность полос в ИК-спектре пропорциональна квадрату изменения дипольного момента[15][16].
ИК-спектрометры[править | править вики-текст]
Основная статья: Инфракрасный спектрометр
Дисперсионные ИК-спектрометры[править | править вики-текст]
В дисперсионных ИК-спектрометрах роль монохроматора может выполнять призма либо — в более новых моделях приборов — дифракционная решётка. Обычно в оптической схеме монохроматор располагается после кюветы с анализируемым веществом, то есть в спектр разлагается излучение, взаимодействовавшее с образцом. При этом последовательно для каждой длины волны излучения регистрируется интенсивность излучения, что и даёт спектр поглощения. На пути излучения установлена щель регулируемой ширины, позволяющая выделить для работы определённый спектральный интервал (обычно от 20 до 0,5 см–1)[17].
Наиболее часто используются двухлучевые дисперсионные ИК-спектрометры. В этом случае излучение источника делится на две части, одна из которых пропускается через анализируемый образец, а вторая — через образец сравнения (чистый растворитель, или таблетка бромида калия без пробы). Эти два пучка попеременно попадают на детектор, где создают сигналы разной интенсивности. Их соотношение даёт величину пропускания Т[17].
Спектрометры с преобразованием Фурье[править | править вики-текст]
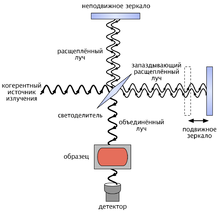
Оптическая схема ИК-спектрометра с преобразованием Фурье
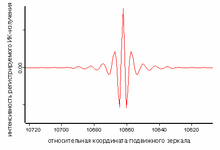
Интерферограмма полихроматического излучения
Главным компонентом Фурье-ИК-спектрометров является интерферометр Майкельсона, известный с начала XX века. Его ключевыми элементами являются три зеркала. Светоделительное зеркало делит пучок излучения на две части, одна из которых отражается от неподвижного зеркала, а вторая — от подвижного (сканера). Оба отражённых пучка затем снова попадают на светоделительное зеркало, где объединяются, проходят через кювету с образцом и направляются на детектор. Подвижное зеркало призвано создавать разницу оптического пути (разность хода) для двух пучков света. При разности хода в (n+½)×λ проходящие пучки взаимно уничтожаются, а отражённые, напротив, усиливаются. В результате получается интерферограмма — график зависимости интенсивности зарегистрированного излучения от разности пути. Для монохроматического света она имеет форму косинусоиды. Для используемого в ИК-спектроскопии полихроматического света она приобретает более сложную форму и содержит всю спектральную информацию о падающем на детектор пучке. Далее интерферограмма пересчитывается в инфракрасный спектр путём преобразования Фурье[18][19].
Преимущество таких приборов заключается в следующем:[20]
-
одновременно регистрируются все длины волн;
-
на детектор попадает более интенсивный поток света за счёт отсутствия щелей;
-
в качестве внутреннего эталона длины волны используется гелий-неоновый лазер;
-
возможна запись спектров в режиме накопления.
Как следствие, значительно сокращается время записи спектра: спектрометры с преобразованием Фурье дают возможность записать до 50 спектров за секунду, в то время как дисперсионный прибор требует около 20 минут для записи одного спектра. Также улучшается качество спектров и чувствительность анализа (на 2-3 порядка) за счёт использования режима накопления. Фурье-ИК-спектрометры обычно однолучевые, что делает невозможным запись спектра с образцом сравнения. По этой причине также не удаётся компенсировать «атмосферные» помехи (наличие углекислого газа и воды). Обычно этот недостаток устраняется путём записи двух последовательных спектров с вычитанием спектра образца сравнения из спектра анализируемого образца, однако в последнее время также приобретают популярность двухлучевые приборы[18].
ИК-спектроскопия пропускания[править | править вики-текст]
Органические соединения[править | править вики-текст]
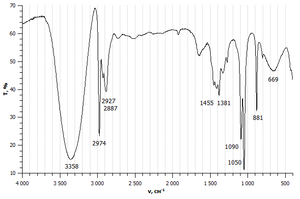
ИК-спектр этанола, записанный из плёнки вещества в режиме пропускания (T)
Колебательные спектры органических соединений обычно имеют сложную структуру и содержат большое число полос разной формы и интенсивности. Экспериментально установлено, что наличие тех или иных полос в определённой области спектра свидетельствует о наличии в молекуле соответствующих им функциональных групп. Однако ни одна группа не является в полной мере изолированной от колебаний остальной части молекулы. Это приводит к некоторым изменениям частоты и интенсивности полос, зависящим от химического окружения функциональной группы[21].
Анализ ИК-спектров многих тысяч органических соединений позволил составить корреляционные таблицы, которые связывают функциональные группы с частотой и интенсивностью колебаний. Однако обычно в спектрах органических соединений присутствуют также полосы поглощения, которые нельзя соотнести с конкретными колебаниями[21].
Колебания связей X–H, где X: C, O или N, можно приближённо описать, как колебания двухатомной молекулы. В этом случае приведённая масса μ всегда близка к 1, а значение силовой постоянной K примерно одинаково для всех подобных связей, поэтому колебания X–H проявляются приблизительно в одной области частот. Например, для связи C–H силовая постоянная равна около 490 Н/м, что даёт частоту в 3000 см–1. Для связей O–H и N–H значение частоты обычно немного выше из-за более высоких значений K[21].