


ББК52.5 Б 90
У ДК 575.167
Рецензенти: завідувач кафедри медичної біології
Буковинської державної медичної академії,
проф. В.П. Пішак, доц. кафедри Т.Є. Дьякова',
директор УНГЦ МОЗ України, д-р. мед. наук,
проф. І.Р. Бариляк; завідувач відділу Інституту
геронтології АМН України, проф. В.П. Войтенко
Бужієвська T.I.
Б 90 Основи медичної генетики. К.: Здоров'я,
2001. - 136 с.
ISBN 5-311-01204-8
Викладені основи медичної генетики, починаючи з визначення понять, законів, історії генетики, молекулярних та хромосомних основ спадковості та закономірностей мінливості організмів. Посібник містить сучасні відомості про методи генетичного обстеження та спадкові хвороби: вади розвитку, захворювання, пов'язані зі спадковими порушеннями різних видів метаболізму, опис фармакогенетичних ензимопатій, деяких первинних імунодефіцитів та онкозахворювань. У ньому наведені сучасні можливості лікування та профілактики спадкової патології, основи медико-генетичного моніторингу, медичні аспекти генної інженерії. Закінчується посібник контрольними питаннями до окремих розділів та задачами з медико-генетичиого консультування, наводиться генетичний глосарій основних термінів та симптомів.
Для студентів-вищих медичних закладів освіти всіх рівнів акредитації та лікарів різного профілю.
![]()
![]()
![]()
![]()
Зміст
|
ВСТУП |
5 |
|
ЗАГАЛЬНА ГЕНЕТИКА |
8 |
|
Історія розвитку генетики та медичної генетики як науки |
8 |
|
Молекулярні основи спадковості |
13 |
|
Хромосомні основи спадковості |
21 |
|
Мінливість живих організмів |
26 |
|
МЕТОДИ ГЕНЕТИЧНОГО ОБСТЕЖЕННЯ |
30 |
|
Клініко-генеалогічиий метод |
30 |
|
Лабораторні медико-генетичні методи |
34 |
|
СПАДКОВІ ХВОРОБИ |
37 |
|
Аномалії та дефекти розвитку |
40 |
|
Порушення метаболізму |
49 |
|
Патологія вуглеводного метаболізму |
50 |
|
Порушення метаболізму амінокислот |
51 |
|
Порушення ліпідного обміну |
53 |
|
Порушення обміну металів |
57 |
|
Порушення інших видів метаболізму |
59 |
|
Фармакогенетичні ензимопатії |
66 |
|
Спадкові хвороби крові, кровотворних органів,дефекти білків плазми |
72 |
|
Спадкові хвороби сечових органів |
76 |
|
Спадкові дефекти неферментних білків |
79 |
|
Первинні імунодефіцити |
82 |
|
Спадковість в онкопатології |
86 |
|
Мітохондріальні хвороби |
89 |
|
Хвороби геномного імпринтингу |
94 |
|
СУЧАСНІ ПРИНЦИПИ І МОЖЛИВОСТІ ЛІКУВАННЯ ТА ПРОФІЛАКТИКИ СПАДКОВОЇ ПАТОЛОГІЇ |
99 |
|
ГЕНЕТИЧНИЙ МОНІТОРИНГ |
103 |
|
МЕДИЧНІ АСПЕКТИ ГЕННО-ІНЖЕНЕРНОЇ БІОТЕХНОЛОГІЇ |
107 |
|
МЕДИКО-ГЕНЕТИЧНЕ КОНСУЛЬТУВАННЯ |
115 |
|
Контрольні питання та задачі |
121 |
|
Генетичний глосарій |
124 |
|
Відповіді на питання та задачі |
130 |
|
СПИСОК ЛІТЕРАТУРИ |
133 |
Вступ
Життя на Землі побудовано за єдиним планом, в його основі лежать єдині механізми і закони, які забезпечують кодування програми особистого існування живої Істоти, передачу цього коду нащадкам, реалізацію про-І рами в конкретних умовах (у просторі та часі) та •Імінення, корегування цієї програми, без чого неможлива сама еволюція. Наука, що вивчає спадковість та мінливість організмів, зветься генетикою. Ця ба-юва біологічна наука має власні цілі: 1. Вивчити .Іакономірності спадковості та мінливості. 2. Використати набуті наукові знання у практичній діяльності людини.
Усі, хто обрав для своєї праці галузь медицини, поза нсяким сумнівом, мають знати генетику загальну, генетику людини, медичну генетику, діагностичні можливості клініко-генеалогічного аналізу, методів молекулярної та біохімічної генетики, цитогенетики. Лікарі будь-якого профілю повинні опанувати основи таких нових галузей науки, як популяційна генетика, імуногенетика, фармакогенетика та онкогенетика, розуміти можливості та принципи медико-генетичного консультування родин. Не володіючи цим матеріалом, лікар не може мислити генетично і тому оцінює норму та патологію, здоров'я та хворобу тільки стосовно вплину шкідливих факторів середовища (інфекційні агенти, порушення у харчуванні, травми, отрути, радіаційне опромінення тощо) на якусь «середньостатистичну» людину. Такий погляд на проблему подібний баченню одним оком плоского зображення замість тривимірної дійсності, пізнання якої можливе тільки за наявності обох очей.
У наш час, наприкінці 20-сторіччя, а тим паче для лікарів наступного віку, обмежене уявлення є не тільки недостатнім, а навіть хибним. «Середньої» людини для медицини взагалі не існує, є конкретна особа певної статі, віку, з індивідуальною, ні на кого не схожою генетичною програмою, що зумовлює саме ЇЇ норму реакції на чинники існування. Двох однакових людей не існує (за винятком монозиготних близнюків). У людини, як і у кожної живої істоти, вся інформація про фізичні, біологічні ознаки, чутливість до навколишнього середовища, схильність до конкретної розумової чи іншоіидіяль-ності, про поведінку, швидкість та силу реакції на зовнішні і внутрішні подразники, несприйнятливість окремих продуктів харчування або лікарських препаратів, надчутливість до інфекцій, радіації, схильність до захворювань різних органів та систем, порушень обміну речовин, нервово-психічної системи та онкопа-тології записана у генетичному матеріалі кожної клітини людського тіла. Всі ж клітини є потемками однієї зиготи — заплідненої сперматозоїдом яйцеклітини. Тобто організм людини — це клон. У момент запліднення об'єднується генетична інформація від батьків і починає реалізовуватися закодована у просторі та часі програма. До миті запліднення інформація в статевих клітинах законсервована, і в їх спадковому матеріалі були сконцентровані стиснуті простір і час. При статевому розмноженні злиття чоловічої та жіночої статевих клітин є пунктом 0 (сигналом «пуск»), стартом для розгортання програми життя організму, починаючи з ембріонального періоду, через дитинство, пубер-тат, дітородний вік, старість, хвороби та смерть. Медична генетика має особливо важливе значення для аку-шерів-гінекологів та педіатрів, які першими зустрічаються з наслідками реалізації певної генетичної про- : грами і мають змогу створювати індивідуалізовані умови для цього процесу з урахуванням його особливостей у кожному випадку. Правильне використання
таких можливостей допоможе у подальшому житті організму не тільки зберегти здоров'я, забезпечити дітонародження, виключити інвалідизацію та передчас-" ну смерть, але й закласти засади для повного вияву кожної особистості, подовження повноцінного життя людини. Без знання генетики неможливо лікувати хворого (а не хворобу), неможливо грамотно, тобто ефективно, попереджувати захворювання, створюючи адаптивне середовище існування для кожної людини. Генетика — це не тільки основа біології, але й філософія життя взагалі та медицини зокрема.
Загальна генетика
ІСТОРІЯ РОЗВИТКУ ГЕНЕТИКИ ТА МЕДИЧНОЇ ГЕНЕТИКИ ЯК НАУКИ
Історія розвитку генетики як науки офіційно починається з 1900 р. і поділяється на 6 періодів:
Тріумфальний хід менделізму.
Утвердження хромосомних основ спадковості.
Відкриття індукованого мутагенезу.
Розвиток біохімічної генетики.
Опанування основ молекулярної генетики.
Становлення мобільної (сучасної) генетики.
До-70 —80-х pp. XX ст. більшість вчених-генетиків вважали, що головні закономірності спадковості та мінливості організмів уже вивчені, тобто генетика як фундаментальна наука досягла своєї першої мети. А тому в майбутньому увага науковців та їх дослідження мають бути скеровані на розвиток генетики різних організмів, у тому числі генетики людини і медичної* генетики та їх прикладних аспектів — біотехнології, розкодування генетичних програм, створення нових гібридних геномів та генотипів, нових живих істот в інтересах «всього людства», окремих популяцій, класів суспільства (навіть груп) або, що рідше, кожної конкретної людини.
В зв'язку з цим проблеми гуманності повністю лягають на плечі лікарів, генетично освічених, які мають стояти на варті здоров'я та життя кожної людини (а не тільки держави або суспільства в цілому), як це ведеться лікарями всіх країн з часів Гіппократа.
Межі історичних періодів розвитку генетики як науки не є остаточно визначеними, вони цілком умовні, бо кожний наступний період зароджувався у надрах
попереднього і мав продовження протягом усіх наступних. Назва ж періодів відображає основний напрямок наукових досліджень саме в цей час.
Головна особливість розвитку генетики полягає в Існуванні величезного доісторичного періоду (протя-Іом усієї історії людства), коли відбувалося накопичення та аналіз факторів спадковості і мінливості ба-і атоклітинних організмів, зокрема й людини. Ознайомлення з пам'ятками культури та релігії свідчить про •Іс, що здобуті людством знання на якийсь час губилися, забувалися, а інколи просто нехтувалися; часто наукові дослідження заборонялися через невігластво або політичні мотиви. Опис родоводів, починаючи з Адама та Єви, що включали кровноспоріднені зв'язки, інбридні ні інцестні шлюби з успадкуванням окремих видів патології, наводиться в Біблії. Поширеність у деяких популяціях осіб-носіїв рідкісних генів, або тих, що мали особливі, відмінні від решти ознаки (лівші та правші гоїцо), також відображена у стародавніх історичних торах.
Перша медико-генетична консультація, що докумен-Іально підтверджена, була подана у Талмуді: не дозволялося робити обрізання плоті хлопчикам, старші браги та дядьки по матері яких страждали на кровотечі. Тут ми маємо не тільки ствердження спадковості гемофілії, але й правильно підкреслюється зчеплене зі статтю рецесивне успадкування цієї патології.
Основні класичні закони спадковості були відкриті/' Г.І. Менделем (рис.1) у 1865 p., хоч існували і працюг Ішли вони завжди, з часу появи життя на нашій планеті. Але потім ці закони з різних на те причин надовго «канули в Лету», аж до 1900 p., коли троє вчених різних країн незалежно один від одного відкрили їх наново: де Фриз (Голландія), Чермак (Австрія) та Корене (Німеччина). Проте закони спадковості назавжди зберегли назву законів Менделя, визначивши окремий напрям у біології — менделізм. Істо-

рія генетики людини фактично вважається наукою, що існує з 1865 р. (а не з 1900 p.), але виходячи не з праці Менделя про закони успадкування якісних, альтернативних ознак, а із праць Гальтона, в яких йдеться про успадкування кількісних ознак. Саме цей рік вважається початком зародження євгенічних уявлень та досліджень про методи й умови впливу на спадкові якості люди-^
її й. Результати проведених досліджень згодом стали :»а основу науки євгеніки, позитивної і негативної, що мгодом переросла у псевдонауку расової гігієни, — расизму та фашизму. Впродовж десятків років людєтво не мало наукового обгрунтування недоцільності, хибності євгенічних уявлень та намагань.
У наш час не викликає сумніву твердження, що євгеніка не має під собою ані філософських чи моральних підстав, ані біологічного обгрунтування, ані генетичної основи. По-перше, при селекції рослин, комах, тварин людина працює у власних інтересах, незважаючи на Інтереси живих істот, яких вважає нижчими за себе. Хто ж буде вирішувати долю людини? Не можна вирішувати долю системи, якщо входиш до складу тієї ж системи. По-друге, ми не знаємо програми майбутньої сполюції людства, які варіанти генів будуть мати селективну перевагу в зміненому нами ж довкіллі, тому не маємо права знищувати гени, що здаються на сьогодні поганими. По-третє, розумова відсталість у пере-иажній більшості є наслідком рецесивної патології, що має клінічний прояв тільки в гомозиготному стані. Такі хнорі передадуть не хвороби своїм нащадкам, а тільки один з двох мутантних генів. Нащадки будуть здоровими як їх бабусі та дідусі. Стерилізація хворих не призведе до зменшення кількості ідіотів, психічно хнорих або злочинців, як це сталося у 30-ті роки в Каліфорнії, де стерилізували 10 000 розумове відсталих.
Кількість хворих та носіїв хвороби регулюється самими популяційними законами (біологічними, генетичними). Соціальні ж закони, юридичні та політичні постанови на це не можуть впливати.
Усім відомі 3 закони Менделя:
1. Одноманітність гібридів першого покоління від схрещування організмів, що стійко відрізняються за аль-
тернативними ознаками: в гетерозиготному стані проявляється домінантний ген.
Розщеплення гібридів другого покоління (схре щування гетерозиготних організмів за якісною альтер нативною ознакою) у співвідношенні 3:1 (три части ни нащадків з проявом домінантної ознаки та одна — рецесивної).
Незалежність комбінування ознак у нащадків батьків, що розрізняються за двома або більше альтер нативними якісними ознаками.
Зумовлене цими законами успадкування називається менделюючим, патологія, що передається нащадкам за законами Менделя, — менделюючою. Таким чином, справедливість наукових обгрунтувань взяла гору. Щиро чи не дуже щиро помиляючись, політичні діячі колишнього СРСР надавали науці, як і моральності, класового змісту, додаючи до так званих буржуазних вчень своє ідеологічне закінчення, тавруючи незрозумілі науки та протиставляючи «менделізм» разом з «вейсма-нізмом-морганізмом» матеріалізму і марксизму-лені-нізму. Саме це стало вагомим підґрунтям для заборони генетичних досліджень у нашій країні, викладання генетики на всіх рівнях освіти, морального та фізичного знищення вчених й спеціалістів у цій царині знання та відповідно — наукової і педагогічної літератури. Організатором і виконавцем антинаукової і антилюдсь-кої політики був Т.Д.Лисенко, а її наслідком стало виховання майже трьох поколінь лікарів, які не знають генетики і відрізняються від медичних працівників інших країн відсутністю генетичного мислення, це позначається на їхньому професіоналізмі.
МОЛЕКУЛЯРНІ ОСНОВИ СПАДКОВОСТІ
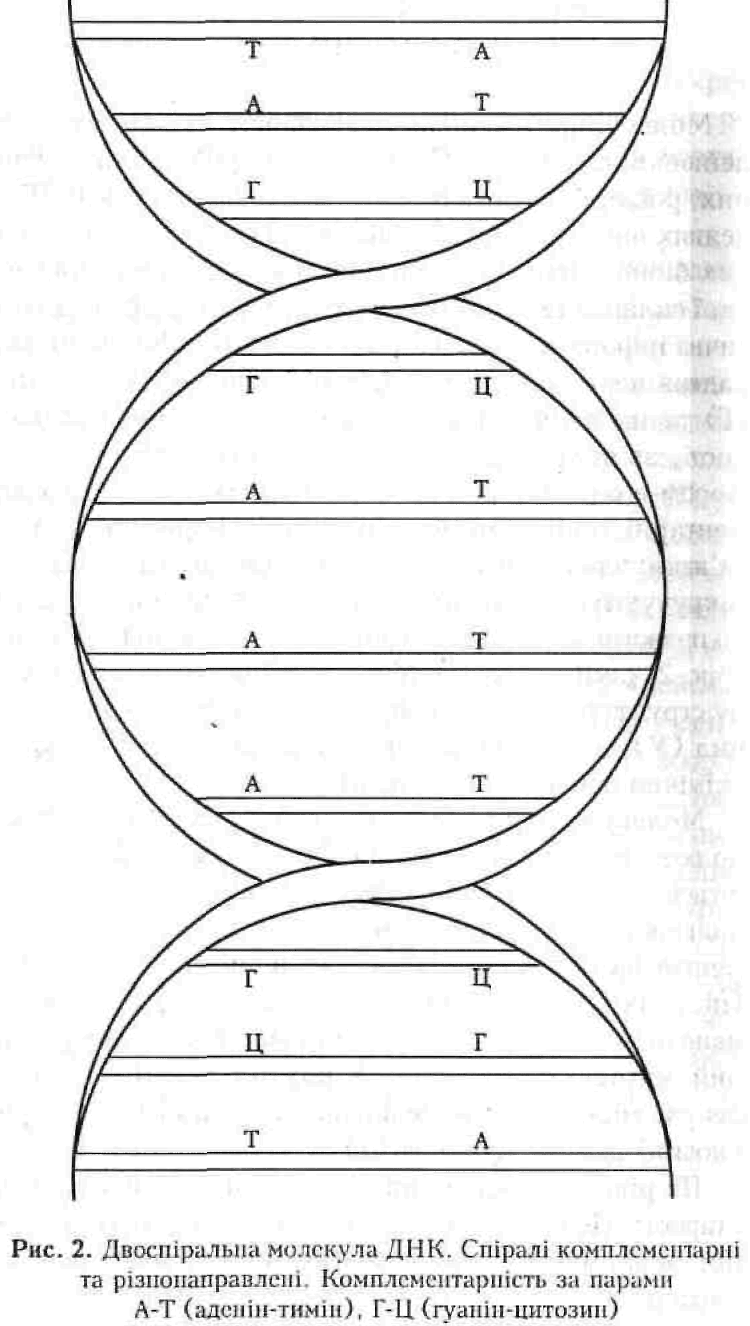
Молекулярні основи спадковості становлять нуклеїнові кислоти — ДНК (у всіх мікробів, одноклітинних, рослинних організмів, комах, тварин) та РНК (у деяких вірусів, зокрема онкогенних). Саме в цих великих біополімерах за допомогою єдиної мови, алфавіт якої складають 4 літери — нуклеозиди, записана генетична інформація живих істот. У ДНК інформація викладена чергуванням аденіну (А), тиміну (Т), гуаніну (Г) та цитозину (Ц), які утворюють певні послідовності, зв'язуючись залишками дезоксирибози та фосфором в одноланцюгову молекулу. Потім два комплементарні один одному ланцюги утворюють водневі зв'язки: аденін-тимін (AT) та гуанін-цитозин (ГЦ), які закручуються й утворюють подвійну спіраль, переважно правогвинтову, одночасно біологічну та інформаційну (рис. 2, «зміїні сходи»). Молекула РНК має односпіраль-ну структуру. До її складу замість тиміну входить урацил (У), а замість залишку дезоксирибози — рибоза (хімічно дещо інша пентоза).
Молекула нуклеїнової кислоти (НК) має здатність до розмноження, подвоєння або реплікації. Розмножуються, тиражуються не білки, а нуклеїнові кислоти. За наявності необхідних компонентів та відповідних ферментів на матриці кожної нитки двоспіральної ДНК (після їх роз'єднання) синтезується комплементарний ланцюг нової ДНК. Реплікація має напівконсерватив-ний, матричний характер. У кожній двоспіральній молекулі міститься і материнський (старий), і дочірній (новий) ланцюг нуклеотидів.
На рівні одноклітинних організмів немає смерті від старості. Цей механізм забезпечує стабільність генетичної інформації, її збереження при процесі передачі нащадкам.
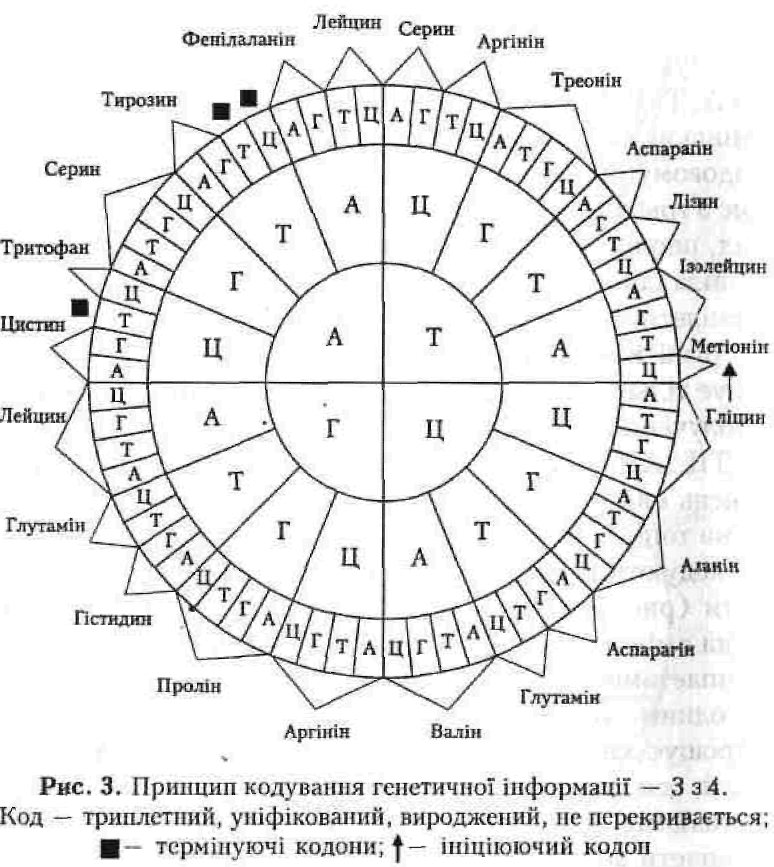
Під час реалізації генетичної інформації відбувається декодування: мова нуклеїнових кислот (чотири літери А, Т, Г, Ц) має бути перекладена на мову білків (20 амінокислот, умовно 20 літер). Це можливо завдяки кодовому принципу: одній амінокислоті відповідає запис з трьох нуклеотидів у нуклеїновій кислоті. Наприклад, послідовність аденін, аденін, аденін (ААА) кодує фенілаланін, а АТТ — лізин. Тому генетичний код — триплетний. Але з 4 літер (А, Т, Г, Ц) можна одержати 64 різні комбінації по 3 літери (43 = 64), а у природі існує тільки 20 амінокислот. Інші триплети (кодони) — сполучення трьох нуклеотидів — не зайві. Три з них (АТЦ, АЦТ, АТТ) — термінуючі, вони свідчать про кінець синтезу, розділові знаки (як у мові — крапка, кома тощо). Інші забезпечують запас міцності геному, бо кодують ті ж самі амінокислоти, що й основні триплети (рис. 3). Тому генетичний код — вироджений: одна амінокислота може бути закодована в ДНК 2—4 триплетами. В одному гені кодони розташовані один за одним, як слова у реченні, не перекриваються, що спрощує запис та робить його стабільним. Генетичний код не перекривається. У всіх живих організмів на Землі в генетичній програмі ті ж самі триплети кодують ті ж самі амінокислоти. Генетичний код ще й у н і ве реальний . Маємо запам'ятати ознаки генетичного коду/ триплетний, вироджений, не перекривається, універсальний. Але у кожному правилі існують винятки. В останні ЗО років дослідники вивчали і збирали такі виключення, їх виявилося багато, виникли нові гіпотези та теорії, що і призвело до виникнення сучасної мобільної генетики, яка прийшла на зміну генетиці класичній. Зараз ми знаємо, що: 1) генетична програма не є зовсім стабільною: існують мобільні дисперговані гени, або елементи, що змінюють своє положення, стрибають з місця на місце; 2) усередині гена існують ділянки зі змістом (екзони) та без нього (інтрони); 3) велика кількість
інформації має регуляторні функції; 4) ген — подільний; 5) у геномі мають місце не тільки унікальні кодуючі послідовності, але й величезна кількість повторів інформації; 6) запис генетичної інформації уюжє відрізнятися від універсального. Інформаційні молекули містяться в клітинах еукаріотів не тільки у ядрі (основна, найбільша програма), але й у деяких органе-лах цитоплазми: мітохондріях, плазмідах, інших ДНК-чи РНК-носіях. Так-дт, в мітохондріях код відрізняється від універсального.
Реалізація"генетичної інформації, а саме синтез білка, здійснюється в цитоплазматичних структурах — ри-
босомах. Для того щоб план будови білка донести від ДНК до рибосом, клітина має спеціальні механізми та рухомі молекули. З того, що знаємо нині, механізм називається транскрипцією, а молекули — це різні види РНК. Транскрипція означає переписування інформації з ДНК на РНК. Головним же в синтезі білка є трансляція — переклад інформації з однієї мови на іншу.
Кодовий запис про структуру білкової молекули переноситься з ДНК на інформаційну (матричну) РНК (вона ж РНК-переносник, лат. «месенджер», синоніми: іРНК, мРНК, т-РНК) шляхом комплементарного, матричного синтезу РНК на ДНК, який можна порівняти з реплікацією (синтез ДНК на ДНК). Молекула РНК копіює весь ген еукаріотів разом з незначущими інтро-нами. Такі тимчасові молекули називаються пре-іРНК.
Молекули пре-іРНК переміщуються з ядра до цитоплазми, а саме до рибосом, що складаються з рибо-сомних РНК (рРНК) та білків. По дорозі пре-іРНК модифікуються, з них видаляються незначущі ділянки кода (інтрони). Значення інтронів, мабуть, дуже важливе, але ще повністю не розшифроване.
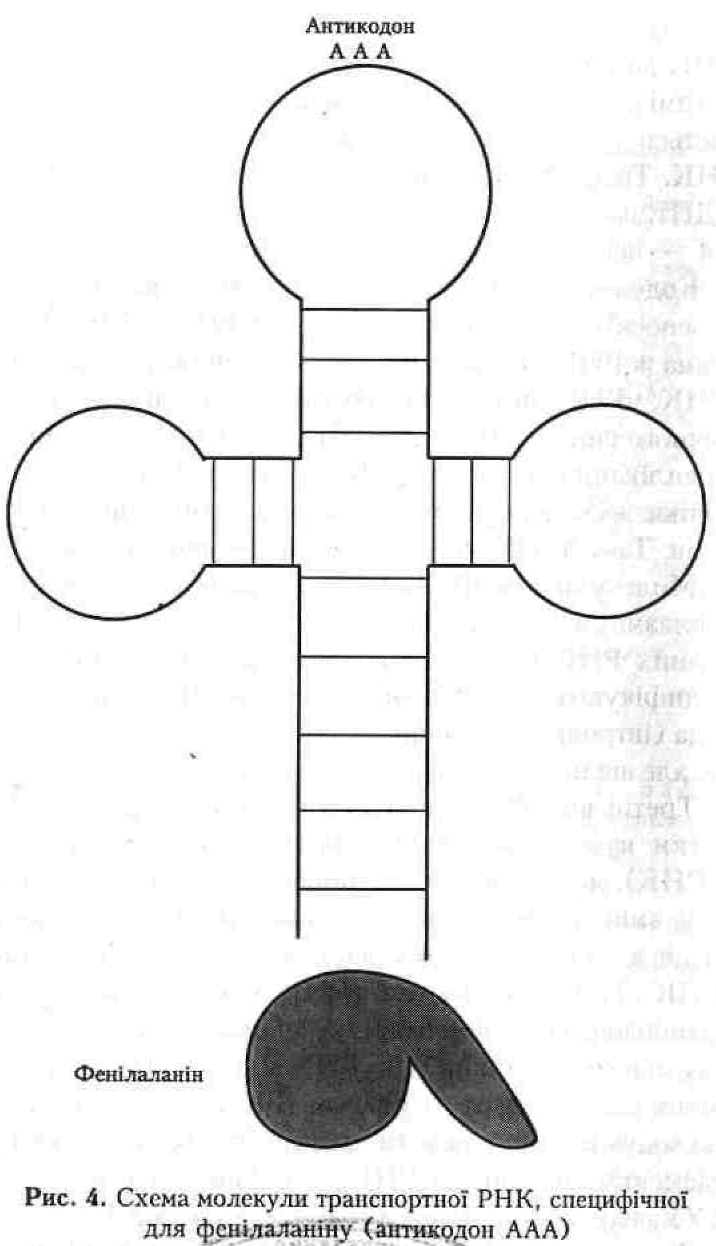
Третій вид РНК складають відносно маленькі (десятки нуклеотидів) молекули транспортних РНК (тРНК), які приносять до рибосом специфічні активовані амінокислоти (рис.4), ставлять їх на відповідне місце в поліпептидному ланцюгу, визначене кодоном іРНК. Тільки молекула тРНК має в своєму складі антикодон, комплементарний до кодону іРНК.
Ми вже визначили, чим РНК відрізняється від ДНК. Білок синтезується за планом іРНК, тому і триплети, що кодують амінокислоти, найчастіше записуються комплементарною мовою РНК, для фенілаланіну це буде УУУ, термінуючі кодони УАГ, УГА, УАА.
Таким чином процес реалізації спадкової інформації від гена до фену (синтез білка — один з них) має вигляд: ДНК>РНК>білок. Довгий час ця формула
_£Z - І потоку генейрНїоІ Інформації вважалася центральною
догмою генетики., s І

У наш час мобільної генетики встановлено існування процесу перенесення інформації від РНК до ДНК. Зворотна транскрипція була передбачена І відкрита С.М Гершензоном (рис.5) та експериментальне остаточно доведена лауреатом Нобелівської премії Г.М.Тьо-
міним. Якщо до цього додати реплікацію ДНК на ДНК та РНК на РНК (можливо, існує у деяких вірусів), то остаточний запис потоку інформації буде мати такий вигляд:

Синтез нуклеїнових кислот на матриці білка поки що не доведений і, напевно, блокований законами термодинаміки. Для його проблематичного існування необхідними мають бути невідомі нам енергопостачальні джерела.
Наприкінці XX ст. стало відомо, що в генотипі людини міститься 50—100 тис. різних генів. Вони кодують продукти, необхідні для існування клітини (кухонні гени), організму (гени розкоші), або, на нашу думку, не кодують нічого. Останні зараз звуться егоїстичними генами, надлишковою генетичною інформацією, що може містити або пам'ять про минулу еволюцію, або бути резервом (планом) майбутньої еволюції.
Увесь обсяг генетичної інформації знаходиться під суворим контролем регуляторних механізмів. Усі гени взаємодіють між собою, створюючи єдину систему. Регуляція їх активності відбувається як за відносно простою схемою — продукт гена змінює активність цього чи іншого гена, так і шляхом складного багаторівневого механізму. Він включає процеси регуляції активності генів на етапах транскрипції (до, під час та після неї), трансляції (до, під час та після неї), узгодженої, каскадної групової регуляції праці генів (їх експресія), участі в цьому процесі гормонів (загальні сигнальні
речовини), хімічної модифікації ДНК та інших загальних модифікаторів експресії генів. Експресія окремого гена залежить від того, в якому складі (генотипі) цей ген перебуває. Тому існує різна пенетрантність (проявлення) та експресивність (вираження) генів як нормальних (дикий тип), так і мутантних алелів.
Ці поняття вперше введені в генетику М.В.Тимо-феєвим-Ресовським. Конкретний генотип людини визначає ступінь пенетрантності та експресивності певних хвороб, навіть до відсутності клінічної картини патології за наявності, здавалося б, необхідної кількості мутантних генів.
ХРОМОСОМНІ ОСНОВИ СПАДКОВОСТІ
Хромосомні основи спадковості були закладені дослідженнями Т.Бовері та В.Сеттена у 1902 р. під час першого етапу розвитку науки генетики, значно раніше, ніж були відкриті молекулярні механізми.
Дослідженнями гістологів та цитологів, які працювали на різних експериментальних моделях, було встановлено, що кожному видові живих істот відповідає власна кількість та будова хромосом, що у більшості живих істот — подвійний (диплоїдний, 2п) комплект хромосом у соматичних клітинах, та що у разі статевого розмноження в істот різної статі склад хромосом однієї пари (статеві хромосоми) різний. Дозрілі статеві клітини мають гаплоїдний (п) набір хромосом внаслідок редукції у мейозі.
Диплоїдність відновлюється під час запліднення, що і призводить до початку нового життя, тобто означає старт, від якого для цієї нової істоти починається відлік часу.
Хромосома — це одна кільцева двоспіральна молекула ДНК, намотана на ланцюжок гістонових бубликів,
внаслідок чого утворюється її 4-рівнева спіралізація. Чим щільніший зв'язок ДНК з гістонами, тим більша спіралізація хромосоми, остання більше інактивована, генетична інформація виключена з процесу. Найвищий ступінь спіралізації хромосом спостерігається під час останніх фаз мітозу (анафаза, телофаза), що зберігає генетичний матеріал від ушкоджень під час його розподілу між дочірніми клітинами. Найменша спіралізація хромосом та порівняно слабкий зв'язок ДНК з гістонами існує у Оо-фазі клітинного циклу, коли клітина здійснює свою диференційовану функцію. У цей період інтерфази експресується основна кількість дієздатних генів, працюють «кухонні» гени — однакові у всіх клітинах організму, які забезпечують власні потреби клітин в РНК, білках, ліпо- та глікопротеїдах, ферментах, енергії та ін. У цей час здійснюється і спеціалізована функція клітин диференційованих тканин та органів, працюють гени «розкоші», продукти яких необхідні всім клітинам організму та виділяються у міжклітинне середовище у вигляді ферментів, гормонів, нейро-пептидів тощо. У фазі G,, в яку входять клітини при підготовці до ділення, починають функціонувати гени, що забезпечують розмноження, у цитоплазмі накопичуються необхідні білки та енергетичні сполуки. Клітина входить у S-період, тобто період синтезу ДНК. До початку S-періоду так звана інтерфазна хромосома має одну хроматиду і по одному плечу з різних боків цен-троміри.
Під час напівконсервативного матричного синтезу утворюються нові дочірні нитки ДНК, що комплементарні материнській нитці; хромосоми одержують другу хроматиду, поєднану в ділянці центроміри з існуючою. У цей час припиняється експресія генів, що працювали у попередні періоди, і хромосома починає швидко спіралізуватися. Через період G2, в якому з'являються структури мітотичного веретена (також внаслідок функціонування відповідних генів), клітина вхо-
дить в мітоз, послідовно проходячи профазу (розпад мембрани ядра, неодночасова спіралізація хромосом), метафазу (коли добре сформовані двохроматидні хромосоми розміщуються по екватору клітини, нанизані центромірою на нитки веретена), анафазу (хромосома розділяється вздовж по центромірі на дві поодинокі хроматиди, що розходяться до різних полюсів веретена) та телофазу (однохроматидні хромосоми починають створювати нові ядра і власні клітинні оболонки кожної нової клітини). Замість однієї материнської клітини виникають дві нові — дочірні, замість однієї старої — дві новонароджені. Доки клітини діляться, вони не вмирають, не старіють, а омолоджуються. На рівні одноклітинних організмів час можна повернути назад.
Кількість хромосом, що містять всі гени організму, постійна дляїкожного виду. Tjio та Levan (1948) вперше визначили, що в кожній клітині тіла людини знаходиться 46 хромосом, тобто для людини є властивим набір хромосом 2п=46, або 23 пари хромосом, з яких одна пара статевих хромосом (XX у жінок і XY у чоловіків) та 22 пари аутосом, однакових у осіб різної статі. Статеві клітини утворюються внаслідок двох послідовних мейотичних поділів клітин гермінативного епітелію, під час яких один із поділів настає без періоду синтезу ДНК (екваційний поділ, або редукція кількості хромосом). Тому кожна дозріла стагева клітина має гаплоїдний, половинний (п) набір хромосом. Сукупність генів, що міститься у хромосомах гаплоїдних клітин, зветься геномом. Усі яйцеклітини мають 22+Х хромосоми, сперматозоїди мають 22+Х хромосоми або 22+Y хромосоми (у співвідношенні 1:1). Стать майбутньої дитини (46, XX або 46, XY) залежить від набору хромосом сперматозоїда, що запліднив яйцеклітину. В той же час яйцеклітина може мати різну вибіркову чутливість до тих чи інших сперматозоїдів. Жіночий організм повністю диплоїдний, чоловічий — гемізигот-
ний за X- та Y-хромосомами. Тобто чоловічі клітини мають всі гени, що розташовані в аутосомах, у подвійній кількості (як і у жінок), а гени, розташовані в статевих хромосомах, у чоловіків існують в одному екземплярі. В результаті статевого розмноження дівчинка отримує кожний ген і від матері, і від батька, а хлопчик отримує гени, що розташовані в Х-хромосомі тільки від матері, а ті, що містяться в Y-хромосомі — тільки від батька. Цей механізм лежить в основі зчепленого зі статтю успадкування. Розміщення генів у хромосомах та їх зчеплене успадкування зумовлює відхилення від закону Менделя про незалежне розщеплення спадкових ознак у нащадків другого покоління (онуки) у разі полі-гібридного схрещування. Це розщеплення обмежується групами зчеплення, кількість яких збігається з гаплоїдним комплектом хромосом (п)+1. У людини таких груп 24=22+X+Y. Y-хромосома значно менша за X і містить менше ДНК, генів, інформації. Різниця в кількості генів у генотипах чоловіків та жінок дещо компенсується спіралізацією в інтерфазових клітинах жіночого організму однієї з двох Х-хромосом. У соматичній клітині жінки працює в більшості випадків одна Х-хромосома, інша ж інактивована, дуже спіралізована і виявляється у вигляді брилки хроматину трикутної, овальної чи округлої форми, що розташована найчастіше біля мембрани ядра. Ця структура має назву хроматин статевий, або тільця Барра (канадський гістолог, який вперше звернув на неї увагу) і використовується для швидкого визначення статі особи, в якої взяли клітини на дослідження, а також для визначення змін кількості Х-хромосом.
Інактивації підлягають різні Х-хромосоми в різних^ клітинах вибірково. В одних клітинах інактивована Х-хромосома материнського походження, в інших — батьківського. Це збільшує мозаїчність жіночого організму, порівняно з чоловічим. Людина — не тільки клон однієї клітини (зиготи), вона — мозаїка у відповід-
ності до роботи різних генів у різних клітинах, що пов'язано з диференційованою функцією поліпотентних клітин багатоклітинного організму. 46 хромосом людини складають її каріотип. Найчастіше каріотип вивчається у період метафази лімфоцитів периферійної крові людини після їх підкультивування поза організмом та спеціального приготування фарбування препаратів (рис. 6).
Наявність у клітинах дишюїдних організмів двох екземплярів кожної хромосоми визначає присутність у них двох екземплярів кожного гена, що розташовані в однакових локусах (ізолокусах) гомологічних хромосом і називаються алелями. Гени бувають алёльни-ми (розташовані в ізолокусах та кодують одну ознаку) та неалельними (різняться за локалізацією, структурою та функцією). Організм, в геномі якого містяться однакові алелі одного гена, зветься гомозиготним за цим геном, а той, що має різні алелі, — гетерозиготним. Деякі гени налічують велику кількість різних варіантів у популяції (кожен організм має тільки два з них). Вони кодують поліморфні білки, що різняться за структурою та функцією. Наприклад, гени, що відповідають за білки еритроцитів та визначають основні групи крові, існують у вигляді трьох різних варіантів: ген A (JA) кодує білок А, ген B(JB) кодує білок В, ген О (J°) взагалі не кодує.
Наявність у генотипі J° J° зумовлює гомозиготність (два однакових алелі) та І (0) групу крові, набір JAJA також засвідчує гомозиготність, але вже II (А) групу крові, яка буде тою ж при гетерозиготності — JA J°. Така ж ситуація можлива у випадку III (В) групи крові: гомозиготний набір JB JB або гетерозиготний — JBJ°. IV (АВ) група крові — завжди наслідок гетерозиготності JA JB. Ген, який кодує альфа-1-інгібітор протеаз, налічує більше 60 різних алелів, а ген глюкозо-6-фосфатдегідрогенази — більш як 100 різновидів.

Що ж таке різні алелі? Звідки вони беруться? Різні варіанти генів спричинюють урізноманітнення живих істот у межах одного виду та є джерелом еволюції, тобто утворюються внаслідок мутаційної мінливості вихідного гена.
МІНЛИВІСТЬ ЖИВИХ ОРГАНІЗМІВ
Мінливість живих організмів може бути спадковою та неспадковою. Неспадкова мінливість за Ч. Дарвіном — •«визначена мінливість» або більш розповсюджена назва — модифікаційна мінливість або просто — модифікації, тобто це мінливість фенотипу організму (що не змінює генотипу) в межах норми реакції, яка закладена у певному генотипі. Модифікації — це і адаптивні зміни, що виникають у чисельних індивідуумів, і у більшості випадків е зворотними, оскільки зникають
після припинення дії чинника, що викликав ці зміни. Вони — специфічні щодо самого чинника. Головний механізм виникнення модифікацій ґрунтується на змінах в регуляції роботи генів. Найбільш сталі серед модифікацій (подовжені у часі) є так звані морфози, які виникають у період ембріогенезу та зберігаються впродовж всього життя організму. Морфози не зникають після видалення чинника, що їх викликав. Вони не мають зворотного розвитку, бо незворотними є стадії онтогенезу та час їх виникнення. До цього виду мінливості належить і тератогенез у людини. Але в деяких випадках онтогенетична мінливість супроводжується модифікацією генетичної програми клітин організму. В наш час починають вводити термін «епігенетична мінливість», яка може бути спадковою.
С.Г. Інге-Вечтомов (1989) розподіляє мінливість організмів таким чином: спадкова — комбінативна та мутаційна; неспадкова — модифікаційна; онтогенетична (подовжені модифікації, тератогенез, морфози), що має риси спадкової та неспадкової мінливості.
Спадкова мінливість — невизначена («спорти») за Ч. Дарвіном — мутаційна мінливість, що виникає внаслідок утворення нових варіантів генетичного матеріалу. Це — зміни в межах гена (алельні варіанти), хромосоми, геному. В зв'язку з цим розрізняються мутації генні, хромосомні, геномні. Такі зміни можуть виникати у соматичних клітинах, що у разі успадкування призведе до клону мутантних клітин тіла; або у статевих, гермінативних клітинах, що може дати мутантних нащадків. Мутації за їх фенотипічним проявом можуть бути летальними, напівлетальними (знижують плідність та життєздатність організмів) або нейтральними і навіть такими, що надають селективної переваги їх носіям за певних умов існування (зростає плідність, життєздатність).
Мутаційна мінливість виникає, також, як і модифікаційна, під впливом факторів довкілля, але тих чинників, що ушкоджують генетичну програму безпосередньо, або опосередковано через ендогенні стресорні механізми. Мутації залежно від причин, Що до них призвели, розподіляються на спонтанні (причина невідома, але обов'язково має бути) та індуковані фізичними, хімічними чи біологічними чинниками. Особливе місце серед біологічних мутагенів займають віруси, живі вірусні вакцини, рекомбінантні ДНК, що використовуються в царині біотехнології, генної інженерії, генної терапії. Такі складники (інформаційні молекули) здатні викликати специфічні (вибіркові) мутації, котрі пов'язані з характером та ступенем комплементарності цих молекул до генетичної програми реципієнта. Такі ДНК- або РНК-носії можуть викликати подовжений мутагенез, епігенетичну спадкову мінливість, діяти як мобільні генетичні елементи, що вибірково інтегруються в геном, або елімінуються, змінюють свою локалізацію тощо.
Фізичні мутагени (УФ-опромінення, радіація) та хімічні мутагени (більш сильні, небезпечні; деякі з них одержали назву супермутагенів) діють неспецифічно, ушкоджуючи мутабельні ділянки геному.
Мутаційна мінливість є основою іншого виду спадкової мінливості — комбінативної, яка забезпечує різноманітність організмів у межах виду та разом з мутаційною створює умови для еволюції. Одним з механізмів комбінативної мінливості є статеве розмноження, за якого виникає перекомбінування генів і хромосом, що містять різні алелі, та відбувається виправлення помилок у програмі під час мейозу (С.М. Гер-шензон, 1996).
Саме в цей період проходять і модифікаційні зміни генетичного матеріалу статевих клітин (епігенетичні), що звуться імпринтингом. Значення та механізми цього процесу ще недостатньо пізнані.
Мінливість організмів необхідна для розвитку життя так само, як спадковість для його збереження.
Знання положень загальної генетики необхідні для розуміння суті медичної генетики, вивчення основних закономірностей виникнення та успадкування патології у людини, створення нових методів діагностики, лікування та профілактики захворювань, виходячи з їх біологічної суті, етіології та глибокого знання патогенетичних механізмів.
Медична генетика — це наука про спадкові хвороби, яка враховує і спадкову схильність. Оскільки генетичні особливості організму існують від моменту запліднення, знання медичної генетики має особливо велике значення для акушерів-гінекологів, неонатологів та педіатрів, які стоять у витоків нового життя. Патологія генетичного матеріалу зумовлює не менше як 50% викиднів, 25% — природжених вад розвитку, 15% — перинатально! смертності, а 10% живонароджених дітей мають спадкові дефекти. Частка спадкових патологій серед усіх хвороб людства постійно збільшується, з одного боку — завдяки успіхам у боротьбі з інфекціями, токсичними пошкодженнями та травматизмом, а з іншого — через поглиблення наших знань у сфері генетики. У наш час можна впевнено сказати, що немає патології, у розвитку якої не брала б участь спадковість, доречі, як і немає такої норми чи такого здоров'я.
Методи генетичного обстеження
КЛІНІКО-ГЕНЕАЛОГІЧНИЙ МЕТОД
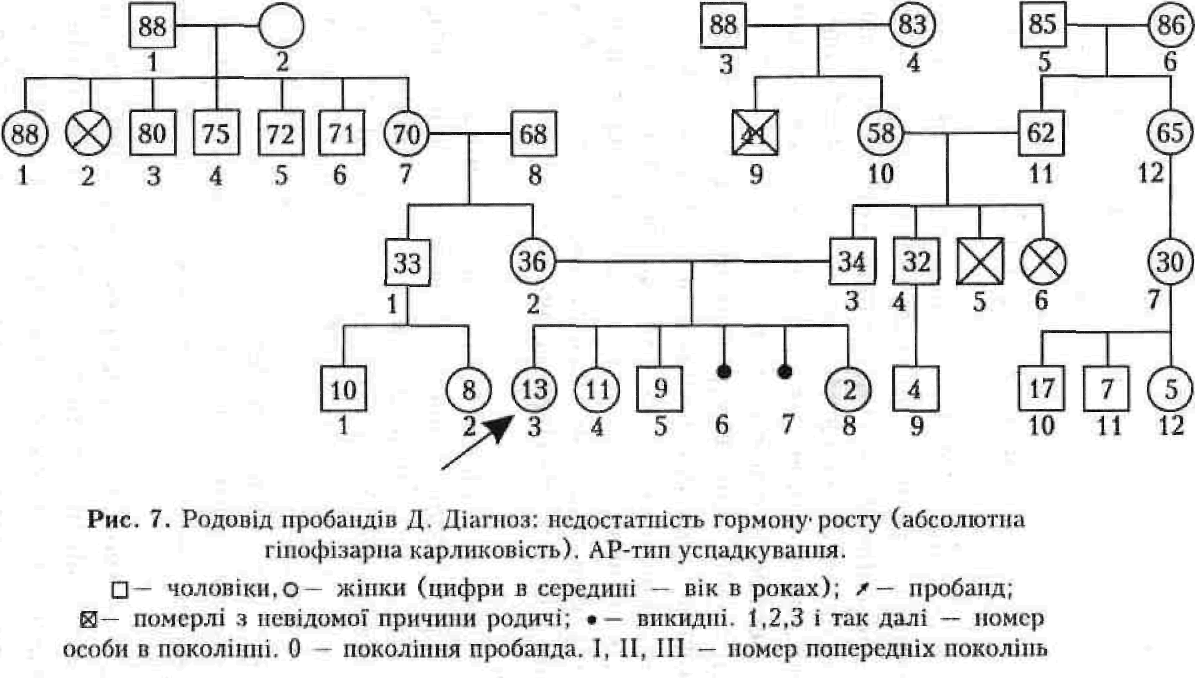
Клініко-генеалогічний метод є головним важелем у медичній генетиці. Більшість діагнозів спадкової патології можна встановити саме за допомогою цього методу аналізу. Він вимагає повного та уважного обстеження хворого, цілеспрямованого збирання анамнезу (про репродукційну функцію та наявність аналогічних уражень серед членів ядерної родини та ряду поколінь за материнською й батьківською лініями), а також клінічного обстеження та інтерв'ювання усіх можливих для цього родичів пробанда (рис.7).
Родовід має бути вивченим за вертикаллю (від покоління до покоління) та за горизонталлю (в межах окремих поколінь).
На основі медичного висновку вимальовується фенотип пробанда за особливою схемою, що відрізняється від записів в історії хвороби, зроблених лікарями не генетиками. Опис фенотипу починається з оцінки поведінки, фізичного розвитку (зріст, маса тіла), контакт-ності хворого, його психічного стану та розумового розвитку залежно від віку. Далі лікар має звернути увагу та відмітити особливості зовнішності пробанда: форма черепа, ріст волосся, його структура і розміщення; форма та розташування вушних раковин (насічки на мочці вуха), брів, очних щілин та відстань між ними. Треба описати форму лоба, носа, ротової щілини, губів, язика, нижньої та верхньої щелеп; наявність, кількість, особливості форми і росту зубів; тверде піднебіння, щілини губи та (чи) піднебіння; форму та розміри
шиї, грудної клітки, хребта. Необхідно також детально обстежити верхні та нижні кінцівки, описати їх форму, кількість пальців, дерматогліфічні особливості (рисунки) долоней та підошв, позиції долоней, підошв.
Потому увага лікаря приділяється стану шкіри: відмічається її еластичність (чи навпаки), ріст волосся, вологість, пігментація, наявність атипових складок, висипання.
Усі відмічені симптоми відповідно записуються спеціальними термінами, що наведені у розділі «Генетичний глосарій». Можливість встановити точний діагноз великою мірою залежить від того, наскільки повно виявлені мікроаномалії розвитку, на які лікарі зазвичай не звертають належної уваги.
Дослідження стану внутрішніх органів традиційними методами пальпації, перкусії та аускультації завершують клінічне обстеження пробанда. З анамнезу принципового значення набувають відомості про перебіг вагітності, строк пологів, маса та зріст дитини при народженні, акушерсько-гінекологічний анамнез матері та родини.
Опитування і клінічне обстеження якомога більшої кількості родичів пробанда має на меті виявлення носіїв патологічних генів як рецесивних, так і домінантних, втім і зчеплених зі статтю. Рецесивні алелі клінічно проявляються тільки в гомозиготному стані (має значення спорідненість батьків), домінантні — як в гомозиготному, так і в гетерозиготному наборі.
Разом з тим наявність та ступінь виразності клінічної симптоматики залежить від пенетрантності та експресивності певного гена.
Пенетрантність — частота або ймовірність прояву будь-якого домінантного гена, вона позначається процентним відношенням кількості осіб, у яких ген проявляється у фенотипі, до всіх носіїв цього гена. Експресивність — ступінь фенотипового прояву гена, міра сили гена, що визначається ступенем розвитку ознаки.
У медичній генетиці термін «експресивність» використовується стосовно повноти виявлення синдрому, а не тільки певного симптому, бо саме синдром, а не симптом є наслідком однієї мутації (при менделюючих хворобах). Часто експресивність моногенного синдрому залежить від статі пробанда. Результатом клініко-генеалогічного обстеження родини є запис фенотипу, родовід, визначення типу успадкування патології та попередній діагноз. Під час складання родоводу використовуються графічні символи. У родоводі (генеалогічному дереві) покоління позначаються римськими цифрами, починаючи з покоління пробанда (0), а кожна людина в одному поколінні — арабськими цифрами. Особа (пробанд), що звернулася до лікаря, відмічається стрілкою. Таким чином, кожна особа з обстеженої родини має свій номер, що відбиває його місце у родоводі (див. рис. 7).
Після встановлення на І етапі попереднього діагнозу (на основі клініко-генеалогічного аналізу) необхідно провести диференційовану діагностику з подібними за клінікою синдромами, генокопіями та фенокопі-ями цієї патології, з метою верифікації діагнозу. При цьому широко застосовуються консультації відповідних спеціалістів: офтальмолога, невропатолога, психіатра, кардіолога, ортопеда та інших, склад яких визначається в кожному окремому випадку. Такому пробан--ду та членам родини призначаються лабораторні та апаратурні дослідження: рентгенографія черепа, кінцівок, хребта (встановлення кісткового віку, виявлення аномалій), ультразвукове дослідження, загальні аналізи крові і сечі, функціональне та лабораторне обстеження серцево-судинної, травної, дихальної, імунної, ендокринної, сечо-статевої та центрально-нервової систем. І знову ж обсяг та комплекс цих обстежень у кожному випадку ґрунтується на конкретних гіпотезах щодо діагнозу, наявності — відсутності змін, що характерні для підозрюваної патології. Ці дії складуть II
етап медико-генетичного обстеження. У деяких випадках вирішальним може бути результат аналізу ендокринного статусу хворого, в інших — імунного статусу чи гістологічного дослідження біопсійного матеріалу. Встановити діагноз в родинах, що раніше мали випадки дитячої смертності істотно допоможуть протоколи розтину.
Більшість спадкових синдромів зустрічається в популяції дуже рідко (ІхІО3 — ІхІО6), тому для верифікації діагнозу слід порівнювати конкретний випадок, з описаними в літературі (атласи, монографії, каталоги, комп'ютерні діагностичні програми, наприклад, «Possum»). У разі спадкової патології точність діагнозу визначає не тільки тактику лікування хворого, прогноз його життя та перебігу хвороби, але й успішність медико-генетичного консультування як хворого (формування адаптивного середовища, вибір професії, подружньої пари, прогноз дітонародження), так і членів його родини — вчасне виявлення уражених індивідуумів, пренатальна діагностика, профілактичне лікування, преконцепційна профілактика, ступінь ризику народження хворих дітей.
Вищевикладене дозволяє зробити висновок: діагностика спадкової патології, успішність її лікування та попередження залежить передусім від компетентності, освіченості лікаря, володіння генетичним мисленням більшою мірою, ніж від наявності дорогої апаратури, реактивів, спеціалізованих установ тощо.
ЛАБОРАТОРНІ МЕДИКО-ГЕНЕТИЧНІ МЕТОДИ
Лабораторні медико-генетичпі методи обстеження мають високу чутливість, що дозволяє однозначно визначити діагноз, характер та локалізацію спадкового дефекту на рівні продукту гена, мутації в гені, хромосомі, геномі.
Ці методи незамінні при масових скринінгах патології, пренатальній інвазивній діагностиці, виявленні носія патологічного алеля, встановленні батьківства, доклінічній діагностиці патології.
У даному посібнику ми обмежимося тільки тим, що коротко перерахуємо ці методи, тому що детальне їх викладення — тема спеціальних монографій та підручників, якими користуються лікарі лаборанти-ге-нетики.
Цитогенетичний метод використовується для дослідження кількості та якості структури хромосом, виявлення хромосомної патології, мозаїцизма, встановлення носіїв збалансованої хромосомної аномалії. До різновидів цього методу відносять різні способи диференційованого фарбування хромосом, з використанням лю-мінісцентних та радіоактивних маркерів, молекулярно-цитогенетичні методи (гібридизація in situ, FISH-ме-тод тощо).
Завдяки застосуванню цього методу все більше па-тологій розвитку людини переміщується з категорії незнаних, спорадичних хвороб до групи хромосомних та успадкованих.
Спеціальні біохімічні методи скеровані на виявлення специфічних продуктів порушеної роботи генів — вільних амінокислот, ліпідів, глікопротеїдів, вуглеводів, ферментів та їх інгібіторів, хибних метаболітів в крові, сечі, тканинах та клітинах організму, амнютичній рідині, хоріоні тощо, а також мінерального дисбалансу.
Дерматогліфічний метод полягає у вивченні малюнків з гребінців, ліній та складок шкіри на долонях та підошвах, а також на долоневих поверхнях пальців. Цим методом досконало володіють деякі ясновидці, віщуни, цигани, але поки що він мало відомий лікарям. Разом з тим одночасне формування рисунків на шкірі вказаних частин тіла та розвиток мозку в ембріональному періоді дає підстави вважати, що дерматогліфіч-
ний метод має значні потенційні можливості, які ще чекають на широке застосування в медичній практиці.
Мікробіологічні методи застосовуються для виявлення продуктів метаболізму в матеріалі від хворих шляхом реєстрації здатності до розмноження в їх присутності залежних мікроорганізмів (ауксотрофів), як у випадку фенілкетонурії.
Молекулярно-генетичні методи дозволяють поставити діагноз носійства певного алелю (в тому числі мутантного) на рівні кодового домену в ДНК, навіть за відсутності продукту експресії гена, визначити гомозиготний чи гетерозиготний стан останнього. Суттю цих методів є використання специфічності роботи ферментів, що розрізають нуклеїнові кислоти, принципу комплементарності нуклеотидних послідовностей та вибіркового розмноження (реплікація, ампліфікація) дволанцюгових ділянок ДНК, здатності забудовувати відсутні ділянки цих структур в умовах пробірки, а не клітини чи організму. Саме на вказану групу покладаються сподівання щодо вирішення проблем пренатальної, доімплантаційної і навіть преконцепційної діагностики, встановлення батьківства або особи злочинця тощо. Ці специфічні, універсальні методи можуть використовуватися в практиці охорони здоров'я, за умови достатнього фінансування. Єдиним недоліком молекулярно-генетичних методів є те, що вони дуже дорого коштують.
У царинах генетики людини та медичної генетики використовуються також популяційні, варіаційно-статистичні методи, метод досліджень на близнюках, які дають можливість вивчати роль спадковості та середовища у розвитку хвороби, розповсюдженні спадкової патології та носійства патологічних алелів в конкретній популяції, встановлювати значення сегрегаційного вантажу і мутаційного тиску.
Спадкові хвороби
Спадкові хвороби — ураження, пов'язані з ушкодженням генетичних структур. При цьому захворювання може успадковуватися від батьків (пробанд-сегре-гант) або вперше виникнути у даного індивідуума внаслідок мутації (пробанд-мутант). І ті й інші здатні успадковуватись у поколіннях (успадковувані хвороби) або не успадковуються у зв'язку з безплідністю ураженої особи.
Спадкові хвороби за характером і широтою ушкодження генетичних структур можуть бути: 1) геномни-ми, тобто пов'язаними з порушенням кількості хромо--сом у всіх клітинах або в певній частині клітин і тканин (мозаїцизм); 2) хромосомними, при яких спостерігається незбалансоване пошкодження структури хромосом типу делеції (втрата ділянки хромосоми), дуплікації (подвоєння ділянки хромосоми), транслокації (перенесення ділянки хромосоми різної величини на невідповідне місце або в невідповідну хромосому) та Інші ушкодження хромосом, котрі спричинюють зміну локалізації та дози генів; 3) моногенними, такими що виникають унаслідок мутації в одному гені, що кодує конкретну ознаку. Серед моногенних хвороб за типом успадкування розрізняють аутосомні й зчеплені із статтю, при яких пошкоджені гени розташовуються відповідно в аутосомах або статевих хромосомах.
Моногенні спадкові хвороби поділяють також на домінантно та рецесивне успадковані. У разі домінантного синдрому мутантний ген проявляється не лише в гомозиготному, але й гетерозиготному стані. При цьому часто аутосомно-домінантні (АД) синдроми летальні в гомозиготі (якщо обидва алелі домінантні та пошкод-
жені). Патологія успадковується дітьми від уражених батьків (вертикальне успадковування в родоводі), а ступінь її тяжкості залежить від пенетрантності та експресивності ушкодженого гена (рис.8).
Х-зчеплена домінантна (ХД) патологія проявляється як у жінок, так і в чоловіків; в останніх ці синдроми перебігають важче, оскільки чоловіки гемізиготні.
Автосомно-рецесивно (АР) успадковувані хвороби уражують дітей, що народилися від здорових батьків — гетерозиготних носіїв мутантного гена — внаслідок комбінативної мінливості. 25% дітей, батьки яких — гетерозиготи, можуть виявитися (згідно законам Менделя) гомозиготами за цим мутантним рецесивним але-лем і бути хворими. Патологія успадковується в родоводі по горизонталі. Усі діти хворих, котрі перебувають не в спорідненому шлюбі, будуть носіями цього гена, але при цьому практично здоровими (див. рис. 7).
Х-зчеплені рецесивні (ХР) хвороби уражають гемі-зиготних чоловіків, успадковуючись від діда через матір (носія мутантного алелю) до онука.
Дівчатка хворіють рідко, якщо від матері й від батька отримують по Х-хромосомі, котра несе рецесивний

мутантний ген. Це траифшрюя при кровноспорідненому шлюбі.
Y-зчеплена патологія передається від батька тільки синові. Вона дуже рідкісна.
АД-патологія здебільшого летальна в гомозиготному стані, може бути наслідком нових мутацій та виявляти мутаційний тиск.
АР- та ХР-хвороби переважно визначають сегрегаційний вантаж і є результатом комбінативної мінливості.
Мультифакторіальна патологія — це наслідок комбінативної мінливості, за якої Діти від обох батьків отри-
мують комплекс мутантних генів, достатній для розвитку патології. Окрім багатьох генів, що спричинюють хвороби, в її етіології й патогенезі велику роль відіграють чинники навколишнього середовища.
Є також хвороби, обмежені статтю, котрі слід відрізняти від зчеплених зі статтю. У цьому випадку мутантні алелі розташовуються в аутосомі (а не в статевій хромосомі), але уражуються тільки особи однієї статі. Наприклад, гіпоспадія розвивається тільки у хлопчиків, а знижена лактація — у жінок.
У даному розділі автор прагне підкреслити роль спадковості в розвитку всіх хвороб людини, а не відокремити лише спадкові, а відтак, дуже рідкісні патології. У зв'язку з цим хромосомні і моногенні синдроми не відмежовано від іншої патології дитячого віку. На відміну від традиційного викладення матеріал подано так, щоб продемонструвати існування спадкової патології зі специфічною етіопатогенетичною природою серед захворювань усіх органів і систем (рис.9). У кожній групі захворювань наводяться лише приклади такої патології, бо тільки моногенних синдромів існує близько 4000, які описані в різних монографіях, атласах, довідниках тощо.
АНОМАЛІЇ ТА ДЕФЕКТИ РОЗВИТКУ
Значні генетичні дефекти геномної природи надзвичайно сильно порушують процес ембріонального розвитку і найчастіше виявляються летальними. Збільшення кількості хромосом, кратне гаплоїдному набору (різні види поліплоїдії), знаходять здебільшого в матеріалі спонтанних абортів. У науковій літературі описані поодинокі випадки народження мертвих або ранньої постнатальної смерті дітей з триплоїдією, тобто коли кількість хромосом — 69 замість 46. На ранніх стадіях
ембріонального розвитку летальні також аномалії кількості окремих хромосом — моносомія та трисомія.
Завершити ембріональний розвиток і народитися живими (у разі геномного рівня ушкодження) можуть діти зі зміненою кількістю статевих хромосом — синдром Шерешевського —Тернера (45, ХО); полісемія X (47,XXX; 48ДХХХ і подібні), синдром Клайнфельтера (47, XXY; 48, XXYY; 48, XXXY тощо), трисомний варіант хвороби Дауна (47, XX (XY) + 21).
Синдром Шерешевського—Тернера і хвороба Дауна проявляються вже у ранньому дитинстві характерним набором численних вад розвитку. Синдром Клайнфельтера найчастіше діагностують у пубертатний період, а полісемію X виявляють здебільшого випадково, і на фенотипі жінок вона позначається далеко не завжди. Зміну кількості хромосом довго вважали новим процесом — наслідком випадкового порушення мейозу в одного з батьків. Такі випадки використовували для моніторингу за темпом мутаційного процесу. Проте є дані, що підтверджують успадкованість факторів, котрі зумовлюють нерозходження хромосом у мейозі. Можлива роль транспозонів (у тому числі інтегрованих у генотип клітин людини вірусних ДНК) як таких, що спричинюють негомологічну кон'югацію, яка призводить до анеуштоїдії (порушення кількості хромосом, некратної гаплоїдному набору) у плода. Серед регулярних трисомій, котрі проявляються від народження, слід зазначити синдром Едвардса (47,XX (XY) +18), синдром Патау (47,XX (XY)+13), трисомію за хромосомою 8 (47,XX (XY) +8).
До хромосомних хвороб, зумовлених порушенням структури хромосом, належать численні природжені вади розвитку, оскільки ця патологія також пов'язана з великими дефектами генетичного апарату, його перебудовами. Перевести деякі вади до цієї групи хвороб з групи синдромів із нез'ясованою етіологією дало
змогу вдосконалити методи цитогенетичного обстеження. На цей час описані синдроми цілковитої трисомії за групами G, Е, Д, часткової трисомії та часткової мо-носомії за всіма хромосомами людини. Для них характерні аномалії розвитку черепа, кінцівок, дефекти розвитку очей, вух, твердого і м'якого піднебень, часто — заяча губа, відставання в розумовому й фізичному розвитку, недоношеність і гіпотрофія при народженні. Поєднання названих аномалій характерні для різних синдромів. Зазначена патологія, що наявна в дитини від перших хвилин життя, а за допомогою пренатальної діагностики реєструється навіть у плода, є показанням до цитогенетичного обстеження. Серед хромосомних аберацій найвідоміші такі: синдром крику кицьки (46, 5р-) та синдром Вольфа —Хіршхорна (46, 4р-). Синдроми котяче око (46, +22q або 47,+ der 22) та Праде-ра —Віллі (46, 15q-) також, за останніми даними, є хромосомною патологією, в етіології якої має значення ге-номний імпринтинг (розділ «Хвороби геномного імпри-тингу»).
Патологія розвитку дитини може мати моно- та полігенну природу. Множинні вади розвитку, успадковувані за АД-типом, наводимо нижче.
Ахондроплазія — карликовість (у зв'язку з укороченням кінцівок) з великим черепом і характерними рентгенологічними ознаками у вигляді вкорочення основи черепа, зменшення потиличного отвору, укорочення та потовщення трубчастих кісток.
Синдром Трічера —Коллінза —Франческетті (манди-булофаціальний дизостоз) характеризується антимон-голоїдним розрізом очей, колобомою, гіпоплазією верхньої та нижньої щелеп, частковою відсутністю вій на нижній повіці, диспластичними вухами, іноді відсутністю зовнішніх слухових ходів, глухотою, щілиною верхнього піднебіння. Тип успадкування — аутосомно-домі-нантний з варіювальною експресивністю.
Синдром Фрімана —Шелдона (обличчя людини, яка свистить) також у більшості випадків успадковується за аутосомно-домінантним типом і відзначається бле-фарофімозом, косоокістю, довгим фільтром (збільшенням відстані між носом і верхньою губою), маленьким ротом з підібраними губами, камптодактилією, клишоногістю.
За аутосомно-домінантним типом успадковується і синдром Крузона, що проявляється гіпертелоризмом, екзофтальмом, розхідною косоокістю, формою носа «дзьоб папуги», короткою верхньою губою, гіпоплазією верхньої щелепи й прогнатизмом нижньої. Іноді відзначається синдактилія, катаракта, застійний сосок зорового нерва, атрофія зорових нервів. Гострота зору звичайно знижується в перші роки життя, потім стабілізується. Нерідко трапляються щілина піднебіння, вади серця.
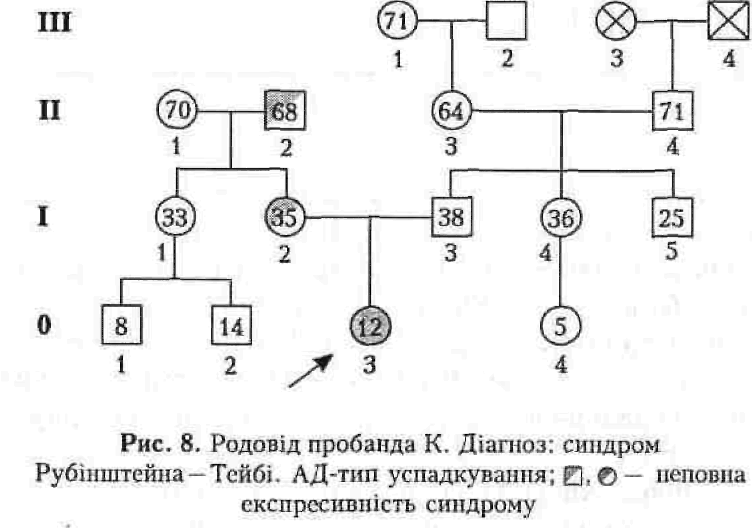
Синдром Рубінштейна — Тейбі (синдром широких перших пальців) проявляється симптомокомплексом: затримка росту, розумова відсталість, розширені кінцеві фаланги перших пальців рук та ступнів, черепно-лицьові дизморфії, гримаса, що нагадує усмішку, крипторхізм, вади розвитку мозку та внутрішніх органів.
Можливим є різний тип успадкування як АР, так і АД з неповною пенетрантністю і експресивністю (рис. 8, 10).
Синдром Опітца (типи І та II) характеризується гіпертелоризмом, щілинами обличчя, вадами розвитку вух, серця; у чоловіків — гіпоспадією, крипторхізмом, паховими грижами.
Синдром Поланда — однобічний дефект великого грудного м'яза з відсутністю соска і дефектами ребер на тому самому боці, а також синдактилія.
Полікістоз нирок (дорослий тип) частіше проявляється після ЗО років. Відзначаються біль у животі, двобічне збільшення нирок, протеїнурія, гематурія. У 50 — 60% хворих спостерігаються артеріальна гіпертен-

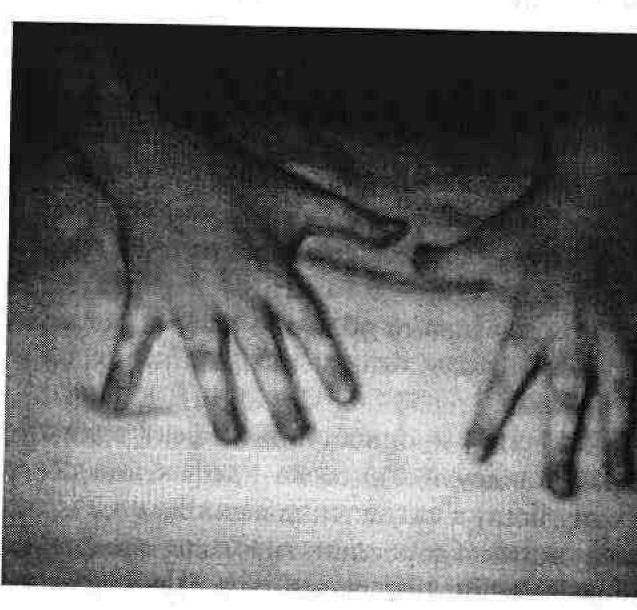
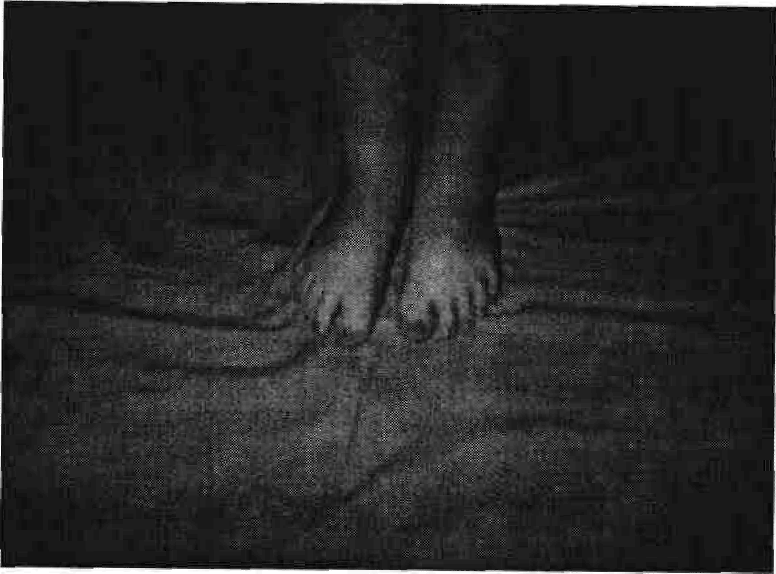
Рис. 10. Синдром Рубінштейна — Тейбі (пробанд 0.2 у родоводі, що на рис. 8). Дівчинка К., 12 років:
а — антимонголоїдний розріз очей, гіпоплазія крил носа, загнутий донизу кінчик носа, гримаса, що нагадує посмішку;
б — широкі кінцеві фаланги пальців рук; . в — широкі кінцеві фаланги першого пальця ніг
зія, прогресуюча ниркова недостатність. Інколи це захворювання виявляють у дітей віком до 10 років.
Синдром Ван дер Вуда окреслений такими ознаками, як щілина губи або піднебіння, ямки на слизовій оболонці нижньої губи.
Синдром Аарськога в осіб чоловічої статі проявляється коротким тулубом, круглим обличчям, гіпертелориз-мом, брахідактилією, шалеподібною калиткою. Вторинні статеві ознаки розвиваються нормально. У жінок відзначаються стерті риси захворювання, у зв'язку з чим нині припускають ХД-тип успадкування цієї патології (жінки-гетерозиготи).
ХД-тип успадкування характерний для синдрому Айкарді, якому властиві одностороння мікроцефалія, асиметричне обличчя, низько розташовані вуха, судоми, специфічна хоріоретинопатія. Шляхом патоморфо-логічного дослідження виявляють агенезію мозолястого тіла.
Серед синдромів чисельних вад розвитку, успадковуваних, за припущенням, аутосомно-рецесивно, необхідно згадати такі.
Синдром Боуена — характеризується черепно-лицьовими й скелетними аномаліями, вродженою глаукомою, вадами серця, відсутністю утворення жиру, агенезією мозолястого тіла, смертю в ранньому віці.
Синдром Сміта —Лемлі —Опітца проявляється гіпотрофією при народженні, мікроцефалією, птозом, епікантом, косоокістю, вивернутими назовні ніздрями, мікрогнатієго, синдактилією, полідактилією, вадами серця, аномаліями нирок і легенів, пілоростенозом, розумовою відсталістю. Нині причиною цього синдрому вважається мутація в одному з генів, порушення функції якого- пов'язане з обміном холестеролу (метаболічна хвороба).
Синдром Гольденгара, якому властива окуло-аури-куло-вертебральна дисплазія, що проявляється від моменту народження однобічною гіпоплазією обличчя, ліпо-дермоїдами очей, колобомою верхніх повік, аномаліями вушних раковин і хребта.
Синдром Жена характеризується асфіксичною дистрофією грудної клітки, зміненою кількістю ребер, відставанням у рості, укороченням кінцівок, нирковою недостатністю, природженою вадою серця.
Мозково-очне-лицево-скелетний синдром проявляється різкою пренатальною гіпоплазією, мікроцефалією, мікрофтальмією, кіфосколіозом, флексорними контрактурами суглобів.
ХР-тип успадкування властивий стенозу сільвієво-го водопроводу, що супроводжується гідроцефалією та
відповідними клінічними симптомами. До синдромів множинних вад розвитку такого типу зарахований і синдром Ленца, що проявляється мікрофтальмією, аномалією пальців, мікроцефалією, деформацією вушних раковин, астенічною ті лобу добою. Успадкування синдрому — Х-зчеплене рецесивне; гетерозиготні жінки мають мінімальні ознаки патології у вигляді вузького обличчя, дефектів зубів і рудиментарної полісиндак-тилії.
Спорадичні випадки множинних вад розвитку належать до патології, пов'язаної з новими мутаціями, оскільки їх успадкування простежити не вдається у зв'язку із значною летальністю. До таких вроджених вад зараховують комплекси амніотичних деформацій (Адам-комплекс, або вроджені ампутації), асоціацію вад VATER: V (vertebrae) — дефекти хребта, A (anal atresia) — неперфорований анус, ТЕ (tracheo-esophageal) — трахеостравоходові фістули, R (radial and renal) — дисплазії променевої кістки й вади нирок. Спорадично трапляється також синдром Кор-нелії де Ланге (мікроцефалія, гіпертрихоз, довгі загнуті вії, брови, що зрослися, довгий фільтр, вивернуті назовні ніздрі, відставання в розумовому і фізичному розвитку, вади внутрішніх органів). У деяких випадках при цьому синдромі описана структурна перебудова хромосоми 3. Можливо, синдром Корнелії де Ланге теж буде включений у групу хромосомних аномалій.
Як спорадичні явища спостерігаються деякі АД-син-дроми (нові мутації), наприклад, ахондроплазія (80% — поодинокі випадки у родині). Ймовірно, що цей відсоток зменшиться, якщо в кожному випадку буде визначатися біологічний батько хворої дитини.
Полігенне мультифакторне успадкування описане для синдрому Поланда (разом з АД-успадкуванням у деяких родинах); при аненцефалії, що супроводжується
гіпоплазією наднирникових залоз і нейрогіпофіза; при атрезії піхви у жінок з нормальним каріотипом (46,ХХ).
Множинні природжені вади розвитку слід відрізняти від аномаладів — комплексів вад, котрі виникають у зв'язку із вторинним проявом головного ураження — первинного дефекту певної тканини або органу, що у процесі ембріонального розвитку спричинює формування решти вад.
У міжнародній програмі моніторингу за вродженими вадами розвитку в теперішній час відсутній показник множинних вад розвитку, бо сучасні, методи дослідження дають можливість ставити діагноз певного синдрому з визначенням хромосомного чи генного дефектів та типу успадкування у кожному розглянутому випадку.
Спадкові вади розвитку мутаційної природи необхідно розрізняти з наслідками тератогенезу, подовженими модифікаціями, що не успадковуються і не передаються наступним поколінням.
Природжені вади розвитку окремих органів і систем належать до складу синдромів або успадковуються самостійно за одним із зазначених типів: АД, ХД, АР, ХР і полігенно. Причиною генетично зумовлених численних природжених вад розвитку і природжених вад окремих органів і систем вважається система Homeobox генів, що їх мутації ведуть до певного порушення ембріогенезу. У наш час ця система всебічно вивчається з метою розробки ефективних методів діагностики даної патології.
Лікування аномалій та дефектів розвитку — хірургічне. Воно дуже ефективне. Деякі дефекти (наприклад, діафрагмальна грижа) можна усунути ще в пренатальний період. Частота виявлення і реєстрації вад розвитку значною мірою залежить від кваліфікації медичних працівників, використовуваних форм статистичне-
го обліку і звітності. Саме повнота реєстрації природжених вад розвитку, а не генетичні ефекти забруднення навколишнього середовища, пояснює різницю їх частоти в різних країнах. Так, частота такої патології на 1000 новонароджених становить: в Угорщині близько 60, у США — 90, а в Україні, за нашими даними, цей показник впродовж 20 років був наближений до 50 із незначними коливаннями по роках.
ПОРУШЕННЯ МЕТАБОЛІЗМУ
Уся патологія обміну речовин, за винятком травматичного або інфекційного ушкодження відповідних органів, має генетичну природу і певний тип успадкування. Залежно від виду гена, функціональних особливостей мутантних алелів і умов існування індивідуума захворювання здатне проявитись у різні періоди життя людини. У разі створення необхідного адаптивного середовища воно може взагалі клінічно не виявитись.
Розрізняють патологію вуглеводного, амінокислотного, жирового і мінерального обмінів. Відповідно до функції ушкодженого гена виділяють ензимопатії, дефекти рецепторних білків, білків-переносників необхідних речовин, дефекти системи виділення продуктів метаболізму тощо. Усі процеси перебувають під жорстким контролем генотипу організму.
Разом з ушкодженням структурних генів, що мають інформацію про будову білка, можливі мутації регуляторних генів (супресори, репресори, модифікатори) або їх регуляторних ділянок (промотори, оператори, активатори, енхансери та ін.).
ПАТОЛОГІЯ ВУГЛЕВОДНОГО МЕТАБОЛІЗМУ
АД-тип успадкування цих захворювань майже не трапляється. Основна маса синдромів успадковується за типом АР (глікогенози, галактоземії, гіпербілірубінемії тощо). До генетичне неоднорідної групи порушень вуглеводного обміну зараховують цукровий діабет.
Аглікогеноз (гіперглікемічні судоми) супроводжується різкою гіпоглікемією, судомами вранці, відсутністю глікогену в печінці. Первинний біохімічний дефект — ушкодження ферментів, відповідальних за синтез глікогену: глікогенсинтетази й уридиндифосфат-глюкозо-глікоген-трансферази. Судомних станів уникають шляхом частих годувань, у тому числі вночі. Аглікогеноз успадковується за АР-типом.
Галактоземія проявляється блюванням, жовтяницею, гепатомегалією, асцитом невдовзі після народження дитини. У подальшому розвивається катаракта, спостерігається затримка росту і розвитку.
За допомогою біохімічного дослідження виявляють галактоземію, зниження активності галактозо-І-фосфат-уридинтрансферази в еритроцитах і тканинах. Галактоземія успадковується також за АР-типом. Діти — гомозиготи за цією мутацією — не можуть вживати молока, а немовлята відмовляються від грудей. Лікування полягає в призначенні дієти (суміші, що не містять молока).
Недостатність галактокинази теж проявляється не-переносимістю галактози. Клінічні симптоми галактоземії та галактозурії такі самі, що й при попередній ензимопатії. Успадковується за АР-типом. Лікування передбачає виключення з раціону молока.
Синдрому Беквіта —Відемана властивий гігантизм плода і гіпоглікемія. Патогенез його незрозумілий. Можливо, він пов'язаний з ензимопатією в межах метаболізму глікогену. Описані всі можливі типи успадкування (переважно АД). Обговорюється роль імпринтингу
в походженні цього синдрому. Проявляється синдром від народження макросомією, макроглосією, пупковою грижею, борозенками на мочках вушних раковин. Спостерігаються гіпоглікемія, гіперліпідемія, гіперхолесте-ринемія, гіпокальціємія. Розвивається помірна розумова відсталість. Лікування включає призначення дієти і хірургічну корекцію вад.
Гіпоглікемія новонароджених відзначається їх гіпертрофією. Такі діти нагадують своїм виглядом тих, хто народився від матерів з цукровим діабетом. Клінічні прояви включають судоми, гіпоглікемію, м'язову гіпотонію. Успадковується це порушення за АР-типом, патогенез його невідомий. Гіпоглікемія ідіопатична, сімейна, проявляється до дворічного віку, причому частіше у хлопчиків. Спостерігаються слабкість, упрівання, підвищений апетит, тремор, судоми, можлива кома. Провокує гіпоглікемію лейцин. Припускають дефекти інсулінази, що сповільнює розпад інсуліну.
Цукровий діабет — генетичне гетерогенна патологія, може бути наслідком аутоімунних процесів, зумовлених порушенням генів з системи HLA, мутаціями в структурній та регуляторній ділянках гена інсуліну, дефектами рецепторів клітинних мембран, наявністю інгібіторів інсуліну в крові, посиленням руйнування інсуліну, іншими генетичними дефектами та різноманітними їх поєднаннями.
У лікуванні даного захворювання великі надії покладені на використання методів біотехнології (генно-інженерний інсулін, пряма корекція гена).
ПОРУШЕННЯ МЕТАБОЛІЗМУ АМІНОКИСЛОТ
У переважній більшості випадків це — аутосомно-рецесивна патологія.
Алкаптонурія — перша з ензимопатій, описаних у людини. Проявляється темною сечею відразу після народження дитини. У подальшому внаслідок накопичен-
ня в тканинах гомогентизинової кислоти темнішають склери й слизові оболонки. У віці 10 — 12 років розвив ваються охронозний артрит і захворювання серцево-судинної системи. Внаслідок генетичного дефекту гомо-гентизиноксидази порушується метаболізм фенілаланіну і тирозину. Успадковується алкаптонурія за АР-ти-пом. Лікування її включає обмеження вмісту фенілаланіну і тирозину в їжі, застосування великих доз аскорбінової кислоти для поліпшення окислювальних процесів.
Фенілкетонурія — порушення метаболізму фенілаланіну на більш ранніх його стадіях. Проявляється в перші місяці життя (найчастіше у віці 6 — 9 міс) затримкою розвитку. У дитини світлі волосся і очі (внаслідок порушення синтезу пігментів), пігментації шкіри немає. Спостерігаються олігофренія, судомний синдром, характерний мишачий запах. Нерідко спостерігаються екзема, дерматит, підвищена чутливість до світла, пастозність, косоокість, ністагм. Захворювання зумовлене відсутністю або зниженням активності ферменту фешлаланін-4-гідроксилази. За допомогою патоморфологічного дослідження виявляється гліозне переродження головного мозку. Успадковується фенілкетонурія за АР-типом. З метою докліні-чного виявлення патології вдаються до масового скри-нінгу новонароджених. За умов раннього призначення дієти, що обмежує споживання фенілаланіну (дієтичні продукти «Берлафен», «Цимогран», «Мінафен», «Апонті»), і суворого лікарського контролю фізичний та психічний розвиток дитини не порушується. Без проведення дієтотерапії в 100% випадків розвивається важка розумова відсталість, ідіотія. Більш як відсоток осіб слов'янського походження є носіями мутантного гена.
Аргінінемія проявляється судомами, блюванням невдовзі після народження дитини. Згодом додаються ге-патоспленомегалія, мікроцефалія, парапарез, відставання в розумовому і фізичному розвитку. У крові й лікворі підвищується концентрація аргініну. Носійство вірусу
папіломи Шоупа, що синтезує аргіназу, призводить до зниження вмісту аргініну в крові. Успадковується аргі-нінемія за АР-типом.
Альбінізм — група захворювань, зумовлених дефектом метаболізму меланіну, котрий утворюється з тирозину. Розрізняють тирозиназопозитивний та тирозина-зонегативний альбінізм (з геморагічним діатезом). Є форми захворювання з глухотою та неушкодженим слухом. Успадковується за АД- , АР-, ХР-типом.
Окрім зазначених порушень амінокислотного обміну, бувають аміноацидемії, пов'язані з дефектами транспортних систем організму, зокрема — з генетичними дефектами сечових органів.
Целіакія — спадкова патологія, що спостерігається досить часто (1:3000). Вона пов'язана з дефіцитом ферментів, які розщеплюють гліодин — частину білка глю-тену, котрий входить до складу пшениці, жита, ячменю, вівса, тобто всіх зернових культур. Патологія проявляється після введення прикорму у вигляді каш, хліба, причому здебільшого наприкінці першого року життя. Внаслідок токсичної дії глютамінпептиду у хворих гине війчастий епітелій кишок. Порушується всмоктування всіх поживних речовин. Спостерігаються ентеропатія, дегідратація, часті випорожнення рідкої консистенції, гіпотрофія. Целіакія успадковується за АР-типом. Лікування полягає у виключенні з харчового раціону каш (манної, вівсяної, перлової), хліба.
У наш час є агліодинові зернові, виведені шляхом селекції сортів, мутантних за відповідним геном. Продукти з таких круп і борошна можна давати дітям, які страждають на целіакію.
ПОРУШЕННЯ ЛІПІДНОГО ОБМІНУ
У цій групі спадкової патології розрізняють хвороби накопичення (внутрішньоклітинне накопичення продуктів метаболізму — сфінголіпідози) та ліпоїдо*
зи, що супроводжуються збільшенням концентрації ліпідів у сироватці крові.
Хворобу Тея —Сакса зараховують до групи ганглі-озидозів (GM2, І тип). Це одна з форм амавротичної ідіотії (рання, дитяча). Характеризується клінічне прогресуючим зниженням гостроти зору, деградацією інтелекту, іншими неврологічними симптомами. У віці 4 — 6 міс до того активна дитина втрачає рухливість, цікавість до навколишньої дійсності, не впізнає батьків, припиняє гратися, сміятися, не фіксує погляду, сліпне. Відзначається гіперреакція на звук (аж до тонічних судом). Дитина помирає через 1,5 — 2 роки від початку захворювання. На очному дні визначається симптом вишневої кісточки. Первинний біохімічний дефект — зниження активності фруктозо-1-фосфаталь до лази гексозамшідази А (можлива пренатальна діагностика явища). Успадковується хвороба Тея —Сакса за АР-ти-пом. Здебільшого хворі й гетерозиготні носії трапляються серед євреїв-ашкеназі. Припускають, що гетерозиготи більш стійкі до туберкульозу. Ефективної терапії захворювання досі немає. Призначають симптоматичне лікування.
ОМ2-гангліозидоз II типу (амавротична ідіопатія Сандхоффа) відрізняється більш злоякісним перебігом, ніж хвороба Тея —Сакса, хоча клінічна картина схожа. Первинний біохімічний дефект — дефіцит гексозамі-нідази А і В. Лікування симптоматичне.
ОМЗ-гангліозидоз II типу (амавротична ідіопатія Нормана —Вуда, природжена форма) розвивається в перші дні життя дитини. Проявляється прогресуючою гідроцефалією або мікроцефалією, судомами, паралічем, сліпотою, затримкою розвитку. Успадковується дане порушення за АР-типом. Ефективної терапії немає, лікування симптоматичне.
Пізня форма амавротичної ідіопатії Куфса (цероїд-ліпофусциноз дорослих) проявляється у віці 15 — 20 років. Спостерігається повільне прогресування органіч-
ної деменції. Зміни з боку органа зору не властиві, проте в окремих випадках виявляють пігментний ретиніт. У термінальній стадії захворювання характерні епілептичні напади, паралічі, нерухомість. До цієї групи захворювань зараховують також GMl-гангліози-доз — тип І. Хвороба Нормана —Ландінга, сімейний нейровісцеральний ліпідоз, характеризується розвитком гепатоспленомегалії після 6-місячного віку, кіфосколіо-зом, брахідактилією, численними згинальними контрактурами суглобів. Звертають на себе увагу такі зовнішні ознаки, як гіпертелоризм, плоске перенісся, низько розташовані вуха, гіпертрофовані ясна. У половини хворих на очному дні відзначається симптом вишневої кісточки. Смерть настає у 2 —3-річному віці. Первинний біохімічний дефект в умовах даної патології — дефіцит бета-галактозидази. Успадковується захворювання за АР-типом, лікування симптоматичне.
Хвороба Дері (ювенільний системний ліпідоз; GM1-гангліозидоз, II тип) характеризується відставанням психомоторного розвитку; спастичним тетрапарезом, судомами, атаксією, м'язовою гіпотонією, що змінюється ригідністю, деменцією. Тривалість життя за такої патології становить 3 — 10 років. Первинний біохімічний дефект — недостатність бета-галактозидази в клітинах, лімфоцитах, сечі. Успадковується за АР-типом. Лікування симптоматичне.
Серед ліпоїдозів, що супроводжуються накопиченням ліпідів у плазмі, слід виділити різні форми гетерогенних патологій типу гіперліпопротеїнемій, гіперлі-підемій, гіперхолестеринемій, тригліцеридемій.
Ліпоїдоз плазматичний І типу (гіперліпідемія жи-роіндукована, ксантоматоз гіперхолестеринемічний, хвороба Бюргера—Грютца) проявляється ксантомами, г&-патоспленомегалією, іноді панкреатитом у віці до 10 років. У крові підвищується рівень холестеролу, триглі-церидів, натщесерце в ній з'являються хіломікрони. В основі патогенезу лежить дефіцит ліпопротешліпази.
Успадковується за АР-типом. Лікування полягає в призначенні безжирової дієти.
Ліпоїдоз плазматичний II типу (сімейна гіперхолес-теролемія, гіпербеталіпопротеїнемія) проявляється з дитячого віку сухожилковим і туберозним ксантомато-зом, атероматозними змінами у вінцевих судинах серця. Ксантоми розташовуються періорбітально, на сідни-цях, сухожилках. У крові значно підвищується рівень холестерину і дещо зростає показник тригліцеридів. Успадковується дана патологія за АД-типом із неповною пенетрантністю. Лікування полягає в призначенні дієти з обмеженням вмісту холестеролу. Щоб знизити його рівень в організмі, застосовують лінетол, деліпін, нікотинову кислоту, холестирамін.
Ліпоїдоз плазматичний III типу у дорослих проявляється бугристими ксантомами, спостерігається також ранній атероматоз. Ксантоми з'являються на стопах. На долонях помітні жовто-оранжеві смуги. У крові виявляють бета-ліпопротеїди дуже низької густини, холес-теролемію, гіперурикемію. Успадковується за АР-типом. Лікування таке саме, як при ліпоїдозі II типу.
Ліпоїдоз плазматичний IV типу (гіперліпідемія ендогенна, тригліцеридемія, індукована вуглеводами) розвивається як у дітей, так і у дорослих. Уражуються вінцеві судини серця. У хворих нормальна перено-сймість жирів, проте знижена толерантність до вуглеводів. У крові значно підвищується рівень тригліцеридів, дещо зростає рівень холестеролу. Успадковується це порушення за АД-типом.
Лікування полягає в обмеженні споживання вуглеводів; рекомендовано вживання олії. Добрий ефект дає призначення лінетолу, деліпіну, нікотинової кислоти, ловастатину, ліпостабілу, есенціале та інших гіполіпі-демічних препаратів.
Ліпоїдоз плазматичний V типу проявляється у віці 20 — 40 років. З'являються біль у животі, панкреатит,
ксантоми, гепатоспленомегалія, гіперурикемія. Значно збільшується рівень тригліцеридів у крові. Описане АД-успадкування з різною експресивністю, проте здебільшого ця патологія поєднується з нефрозом, мікседемою, алкоголізмом тощо. Лікування передбачає призначення дієти, бідної на вуглеводи й жири, а також препаратів, що використовуються при лікуванні інших форм ліпоїдозів.
Останніми роками встановлено зв'язок плазматичних ліпоїдозів з мутаціями генів Аро-А, Аро-В, Аро-С, Аро-Е. На цій основі пропонується ДНК-д і агностика членів сім'ї хворого для доклінічного виявлення носій-ства певної мутації та профілактичного лікування.
Сьогодні особлива увага приділяється генетичним порушенням холестеролового обміну, які супроводжуються зниженням холестеролу в організмі, що може призвести до народження дитини з синдромом Сміта — Лемлі —Опітца (дефект 7-дегідрохолестеролредуктази), до порушень стероїдного обміну, мієлінізації нервової системи, до розумової відсталості та ін. При зниженні синтезу холестеролу пропонують включати в дієту 2 — З курячих жовтка в день. Але ці висновки ще не є остаточними. Дослідження тривають.
Порушення обміну металів
Найбільш відома патологія цієї групи — гепато-лентикулярна дегенерація (хвороба Вільсона —Коновалова) . Це захворювання починається у віці 12 — 20 років. У хворого спостерігається слабкість, біль у животі, жовтяниця, тремор, м'язова ригідність, гіперкінези, дисфагія, дизартрія, псевдобульбарні симптоми. По зовнішньому краю рогівки з'являється кільце зелено-бурого кольору (кільце Кайзера —Флейшера). Розрізняють абдомінальну, ранню ригідно-аритмо-гіперкінетичну, тремтячо-ригідну та екстрапірамідно-коркову форми
захворювання. Абдомінальну форму часто сприймають за вірусний гепатит, що завершується цирозом печінки. Гепатолентикулярна дегенерація супроводжується деменцією, успадковується за АР-типом. Вона пов'язана з мутацією в гені, розташованому на хромосомі 13, котрий бере участь в обміні міді в організмі. Цей дефект призводить до накопичення міді в крові та відкладання цього металу в клітинах печінки, головного мозку, нирок, селезінки, райдужної оболонки, рогівки, в кришталику ока.
Білку церулоплазміну (ген на хромосомі 6), дефект якого вважався раніше головним в патогенезі хвороби Вільсона —Коновалова, зараз приділяється основна увага в порушенні обміну заліза (Fe2—>Fe3) та розвитку анемії і гемосидерозу. Лікування включає призначення дезінтоксикаційної терапії (унітіол, гемодез), пеніцила-міну, купрбнілу або інших хелатів, оксид цинку, тетра-борату натрію. Необхідно виключити з раціону мідьвмісні продукти (мозок, печінка, горіхи, сухофрукти тощо); не можна готувати страву в мідному посуді. Протипоказані вітаміни групи В, жовчогінні, мідьвмісні препарати. За умови правильного і тривалого лікування значно поліпшується клінічна картина.
Генетичний дефект обміну міді зумовлює ще одне важке захворювання — синдром Менкеса, що проявляється судомами, котрі важко припинити, порушенням терморегуляції, жовтяницею, відмовою від їжі, летаргією в період новонародженості. Швидко знижується гострота зору. Волосся дуже кучеряве, ламке, гіпопігмен-товане, виявляються симптоми недосконалого остеоге-незу. В основі захворювання лежить дефект всмоктування і транспорту міді з кишок, що залежить від мідь-вмісного ферменту — цитохромоксидази. У печінці знижується вміст міді, а в сироватці крові — рівень церулоплазміну. Успадковується синдром Менкеса за ХР-ти-пом. Лікування передбачає парентеральне введення солей міді, мідьвмісного гістидину — бажано в анте-
нагальний період, щоб уникнути необоротних змін у головному мозку й кістках. У постнатальний період внутрішньовенно вводять солі міді під постійним контролем її вмісту в сироватці крові.
ПОРУШЕННЯ ІНШИХ ВИДІВ МЕТАБОЛІЗМУ
Спадкова патологія метаболізму мукополісахаридів має назву мукополісахаридоз, що трапляється у вигляді щонайменше 8 самостійних форм. Ця група спадкових захворювань сполучної тканини характеризується ураженням опорно-рухового апарату, внутрішніх органів, очей, нервової системи. Вже через кілька місяців після народження у дитини відзначаються такі зміни: гіпертелоризм, екзофтальм, збільшення черепа, навислий лоб, запале перенісся, широкий ніс, товсті губи (гаргоїлізм). Розвиваються пахові й пупкові грижі. Звертають на себе увагу короткі шия, кінцівки; деформація хребта і грудної клітки, контрактури суглобів. Виявляють також гепатоспленомега-лію, вади серця. Різні форми мукополісахаридозів зумовлені порушенням метаболізму глікозаміноглі-канів на рівні різних ферментів.
Лабораторне дослідження виявляє в сечі підвищену екскрецію глікозаміногліканів (дерматансульфату, кератансульфату, гепарансульфату). При гістохімічному дослідженні відмічається метахромазія фібробластів і лімфоцитів.
Синдром Гурлєра (І тип) проявляється карликовістю, люмбальним горбом, характерною глибокою деменцією, вадами серця. Дитина помирає до 10-річного віку. Основний біохімічний дефект — дефіцит L-ідуроніда-зи. Успадковується патологія за АР-типом.
Синдром Гантера (II тип) характеризується доброякісним перебігом. Інтелект збережений. Тривалість життя досить значна (до 60 років). В основі розвитку
синдрому — біохімічний дефект ідуронатсульфатази. Успадковується за ХР-типом.
Синдром Санфіліппо (III тип) перебіг має відносно легкий. Розвиваються клінічні симптоми мукополіса-харидозу, розумова відсталість. Змінюється поведінка хворого: дитина стає збудженою, агресивною, нездатною зосередитися, порушується сон, погіршується слух. Характерна гетерогенність (типи A,B,C,D) за біохімічними дефектами — ушкоджені різні ферменти. Хворі живуть до 20 — 30 років. Успадкування синдрому Санфіліппо відбувається за АР-типом.
Синдром Моркіо —Брайлсфорда (IV тип) відзначається численними деформаціями скелета, зниженням слуху. У хворого дуже коротка шия, низький зріст. Спостерігають помутніння рогівки, остеопороз, серцева і неврологічна патологія. Тривалість життя — до 20 років. Основний біохімічний дефект — дефіцит галак-тозамін-6-сульфат-сульфатази, або бета-Д-галактозидази. Успадковується дане порушення за АР-типом.
Синдром Шейє (V тип) проявляється грижею та помутнінням рогівки від моменту народження. Інші, помірно виражені, характерні для мукополісахаридозу симптоми розвиваються в шкільному віці. До них відносять тугорухомість кисті, що набуває вигляду кігтистої лапи, ураження аортальних клапанів, погіршення слуху і зору. Біохімічний дефект — дефіцит альфа-L-іду-ронідази (можливо, алельна форма синдрому Гурлє-ра). Успадковується синдром Шейє за АР-типом.
Синдром Марото—Ламі (VI тип) проявляється в 2-3-річному віці. Спостерігається відставання у рості, наростають усі симптоми, властиві мукополісахаридозу. Викривляються гомілки. Хворий помирає у віці до 20 років. Біохімічний дефект — дефіцит арилсульфа-тази В. Успадковується за АР-типом.
Хвороба Слая (VII тип) характеризується такими самими клінічними симптомами, що й синдром Маро-
то —Ламі. Є форми захворювання без гепатоспленоме-галії. Біохімічний дефект — дефіцит бета-Д-глюкуроні-дази. Успадковується також за АР-типом.
Синдром Вінчестера (мукополісахаридоз псевдорев-матичний) проявляється карликовістю, контрактурами, помутнінням рогівки, остео порозом, ревматоїдно подібною деструкцією суглобів. Успадковується за АР-типом.
Лікування всіх мукополісахаридозів симптоматичне: гормональні препарати, великі дози ретинолу, дієта з малим вмістом аскорбінової кислоти. Розробляються методи замісної терапії.
Спадкові порушення пуринового і піримідинового обміну залежно від рівня ушкодження метаболічного ланцюга можуть перебігати або дуже важко з народження — знижується життєздатність, інтелект, з'являються значні неврологічні ураження (синдром Леша — Найгана) або нагадувати хвороби людей похилого віку (подагра).
Синдром Леша — Найгана — природжена патологія, що проявляється спастичним церебральним паралічем, затримкою фізичного і розумового розвитку, аутоагре-сією, а також змінами, характерними для подагри: підвищенням рівня сечової кислоти в крові, гематурією, ури-курією, нефропатією, гострим артритом. Первинний біохімічний дефект — дефіцит гіпоксантин-гуанін-фосфо-рибозил-трансферази. Порушується синтез ДНК. Успадковується синдром Леша —Найгана за ХР-типом. Патогенетичного лікування поки що немає. Нині розробляється пряма генна терапія цієї патології.
Ксантинурія проявляється нирковокам'яною хворобою, у деяких дітей спостерігають нічне нетримання сечі, ускладнення у вигляді пієлонефриту. Знижується рівень сечової кислоти в сироватці крові та екскреція кислоти із сечею. Основний біохімічний дефект — зниження активності ксантиноксидази. Успадковується за АР-типом. Хворим рекомендується більше пити і обмежувати споживання м'ясних страв.
Подагра позначається болем у суглобах, обмеженням їх рухомості та зміною форми. Запальних змін у суглобах немає. Подагра супроводжується нирковокам'яною хворобою, анкілозами. Відзначають порушення обміну сечової кислоти, зниження активності гіпоксан-тин-гуанін-фосфорибозинтрансферази. Гетерогенність генетичної природи захворювання зумовлює форми з АД- і ХР-успадкуванням (алельні синдрому Леша — Найгана), а також із полігенною схильністю.
Адреногенітальний синдром (АГС) — група гетерогенних захворювань, зумовлених генетичними дефектами метаболізму андрогенних гормонів, що належить до спадкових дефектів обміну стероїдів. Описані 5 форм цієї патології залежно від рівня ушкодження біохімічного ланцюга: дефіцит 21-альфа-гідроксилази або 11-бета-гідроксилази, або 17-альфа-гідроксилази, або 20, 22-десмолази, або 3-бета-ол-стероїд-дегідрогенази. Отже, ця ендокринна патологія, як і низка інших спадково зумовлених ендокринопатій, є ензимопатією. Перші два дефекти зустрічаються найчастіше. Захворювання починається в період внутрішньоутробного розвитку.
Адреногенітальний сцндром може бути з втратою солі або без неї. У новонароджених дівчаток спостерігається маскулінізація, гіперпігментація, зниження рівня статевого хроматину при каріотипі 46,XX. Без специфічного гормонального лікування вторинний статевий розвиток не настає.У хлопчиків першим клінічним симптомом є передчасний статевий розвиток, низький зріст. При частковому дефіциті 21-альфа-гідроксилази електролітний баланс не порушується, не змінюється рівень кортизолу і альдостерону. Абсолютний блок ферменту (відсутність 21-альфа-гідроксилази) призводить до розвитку адреногенітального синдрому з втратою солі. Він проявляється блюванням, дегідратацією, тахікардією, гіпонатріємією, гіперкаліємією. У сечі підвищується рівень 17-кетостероїдів і прегнантріолу.
Адреногенітальний синдром з артеріальною гіпертензією. У дівчаток, як і в разі описаної вище форми синд-
рому, спостерігається маскулінізація зовнішніх статевих органів, а у хлопчиків — пігментація калитки. У дитячому віці розвивається артеріальна гіпертензія. Внутрішні статеві органи формуються правильно. Первинним біохімічним дефектом є дефіцит 11-бета-гідроксилази: у сечі підвищується рівень 17-оксикорти-костероїдів і 17-гідроксикортикостероїдів, у крові знижується рівень кортизолу і альдостерону.
АР-тип успадкування властивий обом описаним формам. Призначають відповідну до дефекту замісну гормональну терапію. Хворий повинен постійно перебувати під контролем ендокринолога. Більш повно всі 5 типів АГС та їх різні клінічні форми описані О.А.Беніковою, Т.І.Бужієвською, О.М.Сильванською (1993).
Гіпофізарна карликовість (пангіпопітуїтарна карликовість) пов'язана з недостатністю всіх тропних гормонів гіпофіза і проявляється пропорційною затримкою росту, зниженням тиреотропної та адренокортико-тропної функцій. Можлива повна затримка росту, відсутність статевого дозрівання, прояви різних ступеней олігофренії. Описані спорадичні АР-, ХР- форми успадкування. Застосована терапія — комплексна, гор-монально-замісна — може бути достатньо успішною (рис. 7, 11).
Із спадкових дефектів обміну типу ензимопатій широко відомі порфірії — група захворювань, що спричиняються порушенням синтезу порфіринів і гема. Внаслідок цього в тканинах організму накопичуються різні порфірини, котрі частково виводяться із сечею та калом. Розрізняють еритропоетичну (порушується синтез порфіринів в еритробластах кісткового мозку) і печінкову (порушення синтезу в печінці) порфірію.
Еритропоетичні порфірії (хвороба Гюнтера) поділяють на дві форми — еритропоетичну природжену та еритропоетичну протопорфірію. Захворювання від моменту народження і до 5 років проявляється виразним фото дерматозом (міхурці, пухирі від денного світла і

бактерицидних ламп), кератитом, кон'юнктивітом, гіпер-трихозом, зміною пігментації шкіри, появою в крові нор-мобластів кісткового мозку, збільшенням вмісту уро-порфірину в сечі. Зуби мають червоно-коричневе або фіолетове забарвлення. Відзначається дефіцит уропор-фіриноген-ІП-косинтетази. Характерний АР-тип ус-
падкування. Лікування симптоматичне, у випадку значного гемолізу показана спленектомія.
При еритропоетичній протопорфірії основним симптомом є фотодерматоз, успадковуваний за АД-типом. Лікування полягає в захисті шкіри від сонячного опромінення.
Печінкові порфірії проявляються різними клінічними формами, які зумовлені ушкодженням відповідних ферментів. Описана порфірія гостра переміжна (шведська спадкова), проявляється з дитинства впродовж цілого життя або може бути латентною, або схожою на напади апендициту, але з виразними симптомами ураження ЦНС аж до коматозного стану. Напад гемолізу провокують: прийом лікарських препаратів (барбітурати, сульфаніламіди, дифеніл, гризеофульвін тощо), дія пестицидів,, голодування, інфекції, естрогени (за 2 — 3 дні перед менструацією), прогестерон, пероральні контрацептиви. Характерне АД-успадкування.
Лікування включає призначення дієти багатої на вуглеводи й білки, відміну препаратів, які провокують напад.
Порфірії пістрявій (південноафриканській) властиві фотосенсибілізація (міхурці, пухирі, рубці, пігментація шкіри, гіпертрихоз, ознаки склеродермії) та неврологічні симптоми. Відзначається збільшенням вмісту сігма-амі-нолевулінової кислоти в сечі. Характерним є АД-тип успадкування. Лікування таке ж, як і при переміжній порфірії.
ФАРМАКОГЕНЕТИЧНІ ЕНЗИМОПАТІЇ
Фармакогенетика — наука, яка досліджує генетичну зумовленість реакції на ліки. Якщо лікарський засіб незалежно від дози спричиняє побічні явища типу ферментативних ідіосинкразій, то це є наслідком латентної фармакогенетичної ензимопатії. Ферменти, що метаболі-зують ліки, в організмі проходять дві стадії — прена-
тального розвитку та постнатального визрівання. У зв'язку з незрілістю білоксинтезуючого апарату гепатоцитів у перші місяці після народження кожну генетично здорову дитину слід розглядати як організм, котрий страждає на тимчасову фармакогенетичну ензимопатію. Це необхідно враховувати, призначаючи лікарську терапію дітям у віці до 3 —б міс, особливо в період новонародже-ності, тим паче недоношеним малюкам.
Фармакогенетична ензимопатія може проявлятися незвичайними побічними ефектами, симптомами отруєння, стійкістю різного ступеня до ліків. Подібні явища е результатом мутації гена, що кодує ферменти чи модифікує його дію, або інших порушень в регуляторних ділянках геному. Є рецепторзалежні форми зміненої генетично зумовленої реакції на лікарські засоби. Цю патологію, звичайно, виявляють випадково, в разі призначення певної лікарської терапії. Після визначення у пробанда схильності до побічних, незалежно від дози, реакцій на певний препарат слід цілеспрямовано обстежити всіх його родичів для встановлення прихованої фармакогенетичної ензимопатії.
Серед фармакогенетичних ензимопатій доцільно виділити такі.
Брак N-ацетилтрансферази, що здійснює ацетилю-вання лікарських засобів. Це зумовлює гіперчутливість до гідразиду ізонікотинової кислоти (ГІНК), його повільну інактивацію. Ензимопатія ГІНК призводить до поліневритів, епілептичних нападів. Апресин в умовах цієї патології здатний спричинити важкий стан, що нагадує червоний вовчак. Донор N-ацетилтрансферази — пантотенова кислота, на котру бідна страва новонароджених.
Саме ця обставина і незрілість ферменту зумовлює тимчасову (до 5 міс) фармакогенетичну недостатність у дитини за цим ферментом. Нині роль дефіциту N-ацетилтрансферази в патогенезі цукрового діабету, ви-
разкової хвороби шлунка носить серед науковців дискусійний характер АР-тип успадкування.
Акаталазія — перша описана в літературі фармако-генетична ензимопатія, що характеризується надчутливістю до етанолу і стійкістю до метанолу. Якщо бракує каталази, не відбувається гідроліз перекисів, не функціонує механізм запобігання утворенню метгемоглобіну, під дією окислювачів відбувається гемоліз; підвищується чутливість щодо гамма-променів. В японських і корейських сім'ях з акаталазією пов'язана хвороба Такахара, котра проявляється гангреною тканин порожнини рота, сепсисом. У Німеччині описаний випадок парадонтозу у 4-річної дитини, зумовленого акаталазією. Як заміс-ну терапію за такої патології використовують капсули з каталазою рослинного походження. АР-тип успадкування.
Брак глюкозо-б-фосфатдегідрогенази (Г-6-ФДГ) проявляється гемолітичними кризами у відповідь на введення в організм примахіну, стрептоциду, фенацетину, фенілгідразину тощо. На цю патологію в світі страждають близько 200 млн осіб, здебільшого чоловіки. Ген рецесивний і розташовується в Х-хромосомі, тому патологія успадковується за ХР-типом. Проте продукт гена входить до складу мембрани еритроцитів, що мають клональне походження (з однієї клітини). У жінок відбувається випадкова гіперспіралізація однієї з Х-хро-мосом, і це призводить до гемізиготності за одним з алелів Г-6-ФДГ в різних клітинах гетерозиготних жінок. Якщо більшість еритроцитів походить із стовбурової клітини з активною Х-хромосомою, котра несе мутантний алель гена Г-б-ФДГ, то жінка є чутливою до зазначених вище ліків. У неї спостерігається гемоліз, головний біль, жовтяничність шкіри. Після припинення прийому препаратів пацієнтка одужує.
Особи з браком Г-6-ФДГ чутливі щодо деяких продуктів і нелікарських речовин: конячих бобів (фавізм), червоної смородини, аґрусу, нафталіну. Неонатальна
гіпербілірубінемія певною мірою зумовлена тимчасовим браком зазначеного ферменту.
Брак редуктази глютатіону проявляється при отруєнні грибами й після видалення жовчного міхура. Цей фермент входить до складу мембрани еритроцитів, бере участь у метаболізмі деяких важких металів, талію. Таке порушення може спричинити гіперчутливість щодо талію (назофарингіт, ураження нервової системи, галюцинації, алопеція). Характерний АД-тип успадкування. Ефективність ферменту підвищується при подагрі, цукровому діабеті та у новонароджених.
Брак редуктази метгемоглобіну супроводжується ціанозом, персистенцією HbF, гіперчутливістю до нітратів і нітритів. Тимчасова латентна ензимопатія такого типу найчастіше проявляється у дітей віком до 3 місяців.
За наявності мутантних алелів гена холінестерази люди (переважно ескімоси) чутливі до міорелаксанта дитиліну, введення якого призводить до зупинки дихання і смерті. Стійкість до цього препарату спостерігається при шизофренії.
Виведення деяких ліків з організму пов'язане з обов'язковим їх перетворенням на водорозчинну форму моно- і диглюкуронідів. Це здійснюється за допомогою уридиндифосфатглюкуройової трансферази, що синтезується в печінці, а «працює» в печінці, нирках, головному мозку й слизовій оболонці кишок. За такою схемою з організму виводяться антибіотики (новобіоцин, біоміцин, левоміцетин), сульфаніламіди, барбітурати, опіати, фенацетин, пірамідон, ментол, камфора, фенолфталеїн, білірубін, вітаміни (нікотинова кислота), гормони (естрогени, андрогени, кортикостероїди, трийод-тиронін, адреналін), серотонін, холестерол і деякі амінокислоти. Інгібітор ферменту — прегнандіол обтяжує тимчасову ензимопатію новонароджених за ури-динфосфатглюкуроновою трансферазою. У зв'язку з цим левоміцетин у новонароджених спричиняє синдром Грея: важке отруєння на 3-тю —9-ту добу після зас-
тасування антибіотика, що супроводжується блюванням, напруженням живота, проносом, ціанозом, задишкою, зниженням артеріального тиску. У деяких випадках настає смерть. Відтак призначати левоміцетин новонародженим можна тільки за життєвими показниками у добовій дозі не більше ніж 25 мг на 1 кг маси тіла. Стимуляторами глкжуронового сполучення є вітамін В]2, кордіамін, фенобарбітал. АР-тип успадкування.
Непереносимість лікарських засобів може бути також пов'язана з генетичним дефектом білків плазми крові (переважно альбуміну), що транспортують ліки в органи у зв'язаному стані. Терапевтичну дію справляють тільки вільні лікарські засоби. Зв'язок з альбуміном є формою накопичення, у цьому процесі ліки (сульфаніламіди, саліцилова кислота, синтетичний вітамін К) конкурують з білірубіном. Здатність білків плазми зв'язувати лікарські засоби залежить від віку, на котрий слід зважати в перинатальний період і при лікуванні хворих похилого віку.
Генетичне зумовлена стійкість до лікарських засобів може бути ферментативною (про що йшлося вище) і рецепторною. Остання є результатом мутацій в генах, що кодують специфічні клітинні рецептори або регулюють експресію цих генів (у разі резистентності до атропіну, морфіну, кумаринових протизсідальних препаратів, кальциферолу). Спадкову схильність слід відрізняти від звикання, за якого відбувається індукція ферментів, які метаболізують ліки.
У зв'язку з тимчасовою фармакогенетичною ензи-мопатією новонародженого, а також відповідно до частоти й важкості патологічних реакцій у відповідь на лікарську терапію всі лікарські засоби поділені на З трупи: 1) такі, що показані для застосування в неона-тальний період (бензилпеніцилін, метициліну натрієва сіль, оксациліну натрієва сіль, ампіцилін, еритроміцин,
олеандоміцину фосфат, цефалоридин, цефалексин, кла-форан, ністатин, кофеїн, церукал, фенобарбітал, вікасол, седуксен, натрію оксибутират, пірацетам); 2) такі, що вимагають обережності при застосуванні (атропіну сульфат, аміназин, анальгін, дигоксин, строфантин, Д-пе-ніциламін, теофілін, еуфелін, гентаміцин, амікацин, лінко-міцину гідрохлорид); 3) засоби, протипоказані в неона-тальний період (борна кислота, левоміцетин, тетрацикліни, канаміцин, мономіцин, налідиксова кислота, сульфаніламіди, наркотичні анальгетики — група морфіну).
Наука фармакогенетика виникла в 60-ті роки нашого століття після того, як у частини (близько 20%) вагітних в Європі, яка вживала препарат талідомід (інша назва — софтенон), народжувалися діти з природженими вадами кінцівок (фокомелією). Це спостереження дало науці два уроки: наслідки можна усвідомити тільки через кілька років після введення в масову медичну практику нового препарату; тестування фармакологічних засобів в модельних системах (навіть на ссавцях) не завжди може бути адекватним, бо люди відрізняються за генотипом від твариц. Талідомід в дослідах на тваринах не викликав тератогенезу.
В умовах перенасичення ринку новими лікарськими препаратами треба стежити за їх можливим негативним фармакогенетичним впливом, який може бути різним за частотою від популяції до популяції в зв'язку з неоднаковим генофондом останніх. Особливо уважними треба бути у разі застосування комплексів нових препаратів під час терапії новонароджених та людей похилого віку.
У наш час одними з основних проблем фармакоге-' нетики є визначення ролі фармакогенетичних факторів у виникненні індукованого амідопірином агранулоцитозу, з'ясування зв'язку фармакогенетичних дефектів і .алергій, дослідження впливу генотипу на розвиток алкоголізму, нікотинізму й наркоманії.
СПАДКОВІ ХВОРОБИ КРОВІ, КРОВОТВОРНИХ ОРГАНІВ, ДЕФЕКТИ БІЛКІВ ПЛАЗМИ
Із цієї групи спадкової патології найвідоміші гемоглобінопатії (значно поширені в небезпечних щодо малярії регіонах) та гемофілії. До гемоглобінопатій зараховують такі генетичні дефекти.
Анемія гемолітична, що зумовлена порушенням структури або швидкості синтезу гемоглобіну внаслідок мутацій в генах, що кодують альфа-, бета-, гама- або сігма-ланцюги молекули глобіну, -або в регуляторних ділянках цих генів. Нині відомі понад 200 варіантів таких точкових мутацій. За ступенем важкості клінічних проявів гемоглобінопатії об'єднують у 3 групи: важкі
— з вираженим гемолізом; легкі — зі зтертими клінічними симптомами захворювання; проміжні фор ми. Найпоширеніші аномальні гемоглобіни HbS, HbC, HbE, HbD.
Анемія гемолітична серпоподібноклітинна зумовлена заміною в поліпептиді шостої амінокислоти глутамі-ну на валін (HbS: альфа-2, бета-2, 6 ГЛУ—>ВАЛ) внаслідок мононуклеотидної мутації в ДНК. Клінічний перебіг характеризується гемолітичним кризом, жовтянич-ністю, гепатомегалією, пальці набувають вигляду барабанних палиць, еритроцити — форми серпа. Розвиваються компенсаторна кардіомегалія, гіперплазія кісткового мозку. Успадковується патологія за АР-типом. Під час гемолітичного кризу проводять гемотрансфузії, після його закінчення призначають повноцінну дієту, аспірин, фолієву кислоту.
Анемія гемолітична (НЬС: альфа-2, бета-2, 6 ГЛУ—>ЛІЗ)
— форма, алельна серпоподібноклітинній анемії. Спос терігаються напади гемолізу, біль у животі, геморагіч ний синдром. Визначаються мішенеподібні еритроцити. Спленектомію проводять рідко, необхідності в гемот-
рансфузіях звичайно немає. Захворювання успадковується за АР-типом.
Анемія гемолітична (НЬЕ) — помірна анемія. Вирізняються мікроцитоз і мішенеподібні еритроцити. Характерний АР-тип успадкування. Лікування симптоматичне.
Анемія гемолітична (HbD: альфа-2, бета-2, 121 ГЛУ—> ГЛІА У цьому випадку точкова мутація в гені глобіну призводить до заміни глутаміну в 121 положенні на гліцин. Можливі гемолітичні кризи. Успадкування здійснюється за АР-типом. Лікування симптоматичне.
Зазначені захворювання проявляються клінічне не дише в гомозиготному стані, але й у вигляді компаундів, коли в організмі є 2 різних мутантних алелі одного гена.
Відомі 3 форми гемолітичної анемії, зумовлені фе-: стальним гемоглобіном (HbF). Найпоширеніша анемія [-— вроджена гіпопластична (синдром Даймонда — Блек-фана). Це гетерогенне захворювання, що може успадковуватись або за АД-, або за АР-типом. У 3-місячному віці з'являються блідість, потім тахікардія, серцева недостатність. Збільшуються печінка і селезінка. У зв'язку з частими трансфузіями крові можливий розвиток гемосидерозу.
Окрім гемоглобінопатій, відомі анемії, спричинені мутантними формами білків, котрі входять до складу мебран еритроцитів, у тому числі ферментів. Деякі з цих захворювань описані в розділі фармакогенетич-них ензимопатій.
Гемофілія — це результат генетичних вад білків, причетних до каскадного процесу коагуляції. На даний час визначені 10 спадкових дефектів, котрі призводять до порушення зсідання крові. Найчастіше трапляються гемофілія А (дефектний фактор VIII) і В (дефектний фактор IX). Обидва гени розташовуються на Х-хро-мосомі, відтак успадковується дана патологія за ХР-
типом. Гемофілія передається від діда матір'ю до онука; це було відомо ще до нашої ери й знайшло відображення в записах Талмуду, де йшлося про небезпеку обрізання у хлопчиків, чиї старші брати й дядьки по матері страждали на кровоточивість. Мутація, що відповідає за гемофілію А, трапляється з частотою 1:10 000, гемофілію В — 1:100 000 (у 10 разів рідше). Водночас гемофілія В у популяції спостерігається лише в 5 разів рідше у зв'язку з ранньою смертністю дітей з гемофілією А. У 28% випадків — це прояви нових мутацій.
Гемофілія А, або класична гемофілія, характеризується кровотечами, гемартрозами і здебільшого розпізнається на 2 —3-му році життя, коли дитина починає активно самостійно пересуватися. У важких випадках у хлопчиків одразу після народження виявляють ке-фалогематоми, кровотечу з пупкового канатика. Типові Крововиливи в колінні, ліктьові, гомілкові суглоби, що призводять до їх тугорухомості й деформації. Відзначається зниження прокоагулянтної активності VIII фактора зсідання крові. Популяційна частота становить 1:2500 живонароджених хлопчиків.
Клінічна картина гемофілії В, або хвороби Крістма-са, схожа на описану вище. У крові знижена активність IX фактора, подовжений час зсідання крові.
Розвиток 5% гемофілій пов'язаний не з цими мутаціями, а з появою в організмі аутоантитіл до VIII і IX білкових факторів. Можливе зчеплення з HLA генами. При лікуванні таких хворих необхідно використовувати амінокапронову кислоту. У решті випадків проводять замісну терапію відповідними білковими факторами під контролем їх співвідношення в крові хворого.
Хвороба Віллєбрандта (псевдогемофілія судинна, капіляропатія) характеризується дуже тривалими кровотечами при ін'єкціях, порізах, мікротравмах, а також носовими кровотечами, гіперменореєю у жінок.
Можливі й шлунково-кишкові кровотечі. Остеоартроз звичайно не розвивається. Шляхом лабораторного дослідження визначають подовження часу зсідання крові, зниження прокоагулянтної та антигенної активності фактора VIII, агрегації тромбоцитів у присутності ри-стоцетину. Успадковується патологія за АД-типом. Лікування включає призначення кровоспинних засобів, переливання свіжої плазми, крові. Застосовують також криопротеїни. Тимчасове поліпшення настає при використанні кортикостероїдних гормонів, серотоніну, ад-реноксину та ін. З віком стан здоров'я хворих поліпшується.
До аномалій білків плазми крові зараховані такі патологічні стани.
Анальбумінемія — може проходити безсимптомно, проте іноді супроводжується набряками (альбумін переносить різні речовини й підтримує онкотичний тиск).
Абеталіпопротеїнемія — супроводжується зміною форми еритроцитів, на поверхні яких з'являються остисті виступи, а також стеатореєю в рацньому віці. У більш старших дітей спостерігається атактична нейро-патія, у дорослих — пігментний ретиніт. У плазмі крові різко знижується рівень холестерину, фосфоліпідів і тригліцеридів. Успадкування відбувається за АР-типом.
Анальфаліпопротеїнемія (хвороба Танжьє) — характеризується збільшенням мигдаликів, що набувають оранжевого кольору, печінки, селезінки й лімфатичних вузлів. У крові знижується рівень холестерину і ліпо-протеїдів високої густини. Даній патології властивий АР-тип успадкування.
Відсутність трансферину веде до гемосидерозу і цирозу печінки.
Брак церулоплазміну спричиняє розвиток гемосидерозу та спостерігається при хворобі Вільсона—Коновалова.
СПАДКОВІ ХВОРОБИ СЕЧОВИХ ОРГАНІВ
Окрім вад розвитку сечових органів, котрі належать до синдромів хромосомних хвороб або можуть бути одиничними вродженими вадами розвитку генетичного або тератогенного походження, а також полікісто-зу нирок та нефрологічної симптоматики, що супроводжує майже всі синдроми патології обміну речовин, необхідно виділити спадкові тубулопатії.
Ці порушення бувають у дітей різного віку значно частіше, ніж вважають. Вони залишаються недіагнос-тованими і призводять до інвалідизації та ранньої смерті.
Первинна тубулопатія характеризується важкою ос-теопатією, нефрокальцінозом, нефролітіазом. Головними симптомами цієї патології виступають поліурія, полідипсія, гіпоізостенурія, лужна реакція сечі, різко виражений метаболічний ацидоз, значний дефіцит гідрокарбонатів у крові, зміна іонограми крові (гіпокаліємія, гіпокальціємія, гіпонатріємія, гіпохло-ремія тощо). Такі діти відстають у фізичному розвитку. В основі патогенезу спадкової тубулопатії лежить генетичне зумовлена неспроможність нирок знижувати рН сечі.
Розрізняють 2 типи ураження канальців: 1) проксимальний, що визначає дефект реабсорбції гідрокарбонатів; 2) дистальний, за якого обмежений транспорт Н+ іонів через клітинні мембрани дистальних канальців.
Вторинна гіперфункція прищитовидних залоз зумовлює підвищення рівня фосфатів. Виділяють такі, найбільш часті, форми патології.
Нирковокам'яна хвороба — генетичне зумовлена патологія обміну кальцію. Солі кальцію виводяться із сечею. Розвиваються гіпокальціємія, остеомаляція, уповільнюється окостеніння хрящів, спостерігається їх по-
"товщення. Найсерйознішим симптомом є прихований та явний ацидоз, котрий вимагає корекції. При нирковокам'яній хворобі необхідне введення великої кількості рідини, солей магнію та фосфатів, які поліпшують розчинність солей кальцію. Якщо є набряки, замість NaCl слід застосовувати солі калію. У випадку значної остеомаляції вводять кальцій та кальциферол (до 50 000 ОД на день).
Ацидоз нирковий канальцевий дорослих (синдром Баттлера—Олбрайта) — починається викривленням ніг, болем у кінцівках. Розвиваються нефрокальциноз, пієлонефрит, тубулопатія дистального типу. Відзначається відставання у фізичному розвитку. Успадковується дане порушення за АД- або АР-типом (різні форми).
Ацидоз нирковий канальцевий інфантильний (синдром Лайтвуда) — уперше відзначається у віці 1 — 18 міс, часто у зв'язку з початком прикорму (сир, коров'яче молоко). Симптоми хвороби — апатія, спрага, слабкість, блювання, ексикоз, поліурія, втрата маси тіла, нерідко судоми, гіпокаліємія. Тип успадкування — ХР або частково обмежений статтю (70% хворих — хлопчики). Прогноз сприятливий.
Синдром де Тоні — Дебре — Фанконі (рахіт нирковий, канальцевий) — проявляється в будь-якому віці, частіше на 2-му році життя. Дитина стає млявою, з'являються субфебрилітет, спрага, поліурія, гіпотонія м'язів. Спостерігаються остеопороз, викривлення трубчастих кісток, спонтанні переломи, а також глюкозурія, аміно-ацидурія, фосфатурія. Характерний АД-тип успадкування з різною експресивністю. Зараз цей синдром також розглядають у групі мітохондріальних хвороб.
Синдром Альпорта (нефрит гематуричний, хронічний, спадковий) — починається в дитячому віці, проявляється слабкістю, частими гострими респіраторними інфекціями, що ускладнюються гематурією. Перебіг торпідний, пізні й рідкі симптоми — артеріальна гіпертензія, набряки. Синдром характеризується природже-
ними катарактами^ ністагмом, міопією, а головне — глухотою, сферофакією, переднім лентиконусом, тапеторе-тинальною дегенерацією. Виявляють такі біохімічні зміни, як гіпераміноацидурія та гіпераміноацидемія, або тільки гіпераміноацидурію, або збіднення спектру амінокислот крові й сечі. Розрізняють 6 типів синдрому, що успадковуються за АД-, АР-, ХР-типом. Відкриті мутації в генах колагену. Тубулопатія при цьому синдромі є вторинною.
Глюкозурія ниркова — проявляється виділенням глюкози із сечею (до 50 — 60 г за добу). Концентрація глюкози в крові не змінена, навантажувальні криві нормальні. Розрізняють глюкозурію типу А, за умови якої знижені межа фільтрації глюкози та її реабсорбція, і типу Б, яка супроводжується зниженням межі фільтрації глюкози за нормальної реабсорбції. Описані АД- і АР-типи успадкування.
Нирковий нецукровий діабет — проявляється на З — 6 міс життя значним діурезом, схильністю до закрепів, блюванням, підвищенням температури тіла. Відчуття спраги у маленьких дітей може не бути, проте поліурія різко виражена: діурез у грудної дитини часом досягає 2 л за добу. Розвиваються сольова гарячка, судоми, гіпотрофія. Відзначається відставання у фізичному розвитку. Ниркові канальці не реагують на анти-діуретичний гормон нейрогіпофіза. Дане порушення успадковується за ХР-типом. Лікування симптоматичне. Вдаються до дієтотерапії (обмеження кількості солі й білка в страві, збагачення її жирами та вуглеводами). Необхідно зменшити дегідратацію. Діурез знижують за допомогою гіпотіазиду.
Фосфат-діабет (вітамін-О-резистентний рахіт, синдром Олбрайта —Баттлера —Блюмберга) — проявляється, коли діти починають ходити, наприкінці першого року життя. У дитини викривлюються ноги, з'являється качина хода. У важких випадках малюки не в
змозі ходити. Розвивається гіпофосфатемія, різко підвищується активність лужної фосфатази. Діти відрізняються низьким зростом, у них часті переломи кісток. Захворювання зумовлено зменшенням реабсорбції фосфатів у ниркових канальцях. Аміноацидурії та глюкозурії немає. Характерний ХД-тип успадкування із 100% пенетрантністю.
Лікування полягає в тривалому застосуванні великих доз кальциферолу, при цьому слід контролювати співвідношення кальцію та фосфатів в крові. Добрий ефект дають 1,25-дигідроксикальциферол і тахістерол, що поліпшують всмоктування кальцію в кишках, а також пиття мінеральної води, котра містить неорганічні фосфати. При багатьох спадкових хворобах обміну речовин розвиваються вторинні тубуло"патії.
СПАДКОВІ ДЕФЕКТИ НЕФЕРМЕНТНИХ БІЛКІВ
До цієї групи перш за все треба віднести спадкові нервово-м'язові хвороби, надзвичайно гетерогенні за своєю природою; це міопатії та міодистрофії, що розвиваються внаслідок ушкодження м'язових структур або нервових елементів. Серед прогресуючих м'язових ди-строфій найбільш вивчені Х-зчеплені хвороби, зумовлені різними мутаціями в гені білка дистрофіку.
Міопатія Дюшенна — характерна раннім проявом клінічних симптомів: дитина стає малорухливою, не бере участі в забавах; у віці 3 — 5 років важко піднімається сходами, протягом наступних 5 — 10 років повністю втрачає здатність рухатися. Розвивається псевдогіпертро-фія м'язів гомілок внаслідок заміщення м'язових структур сполучною тканиною, страждають серцеві м'язи. У сироватці крові вже на початку клінічних проявів хвороби зростає рівень активності креатинкінази та кре-атинфосфокінази. Але це — вторинна біохімічна ознака, яка свідчить про порушення мембран м'язових клітин.
Посилення клінічних проявів міодистрофії, погіршення стану хворого супроводжується зниженням рівня активності креатинкінази в крові. Тип успадкування — ХР. Можлива ДНК-діагностика цієї патології і на її основі пренатальна діагностика (в тканинах плоду).
Міопатія Беккера — відрізняється від попередньої форми клінічним проявом у віці 20-30 років, подовженим доброякісним (без втрати рухомості та ураження серцевих м'язів) перебігом, псевдодистрофія менш виражена, ніж при міопатії Дюшенна, слабкішими є й біохімічні порушення. В основі цієї спадкової хвороби лежить мутація того ж гена дистрофіку, яка розташована в іншій його частині і коротша. Тобто міопатії Дюшенна і Беккера — це різні алельні форми генетичної патології. Тип успадкування — ХР. Можлива ДНК-діагностика та пренатальне встановлення діагнозу.
Лікування міопатій поки що має симптоматичний характер, скеровується на покращання трофіки м'язів (комплекси амінокислот, АТФ, вітамінів групи В та Е, антихолінестеразні препарати, гемотрансфузії, місцева стимулююча терапія).
Муковісцидоз або фіброз кістозний (підшлункової залози) — спадкова хвороба, що зумовлена мутацією в гені трансмембранного регуляторного білка, який відповідає за транспорт натрію, хлору та води крізь мембрани клітин. Вперше в історії за допомогою методів молекулярної біології (зворотна генетика) був відкритий та локалізований ген на хромосомі 7q 31.1, визначені та розшифровані численні мутації в цьому гені, які у гомозиготному стані або у вигляді компаундів (2 різних мутантних алелів ) викликають захворювання. У той же час залишався невідомим первинний біохімічний дефект, тобто продукт експресії цього гена. Хвороба розповсюджена з частотою 1: 2000 — 2500 новонароджених. Клінічний перебіг захворювання може починатися з періоду новонародженості, мати дуже тяжкий симптомокомплекс так званого меконіального ілеуса,
коли густа та в'язка пробка з меконію викликає кишкову непрохідність і всі наслідки цього процесу. В подальшому в перші 6 міс життя дитини розвивається клінічна картина муковісцидоза, в якій можуть мати перевагу спонтанні ураження легенів (легенева форма), кишкового тракту (кишкова форма) або змішана симптоматика. При всіх формах хвороби порушені секреторні процеси у відповідних клітинах всіх екзокринних залоз. Хворі важко дихають, у них спостерігаються коклюшеподібний спазматичний кашель, хронічні бронхолегеневі запалення, емфізема, ателектаз легенів. Після введення прикорму з'являються часті дефекації з великою кількістю світлих калових мас, маслянистих з неприємним запахом, збільшується печінка, розвиваються цироз печінки, гіпотрофія, кахексія. Якщо неправильно лікувати хворих, вони вмирають у віці до 10—12 років. Хворі на муковісцидоз діти добре інтелектуально розвинуті. За умов постійного відповідного лікування хворі живуть навіть до 40 років. Специфічне діагностичне значення серед лабораторних показників має підвищення концентрації натрію та хлору у поті, нігтях та волоссі («солона дитина»,' «гіркі сльози»). У 3 —б-річних дітей у вмісті дванадцятипалої кишки виявляється зниження активності ліпази, діастази, трипсину. Можлива ДНК-діагностика, пренатальна діагностика. АР-тип успадкування. Лікування: дієтотерапія (калорійна, білкова, солона їжа, бідна на жири); ферментотерапія (креон, вобензим, панкреатин та подібні), інгаляції з муколітиками, вібраційний масаж для полегшення виділення слизу, анаболічні гормони. Під час інфекційних ускладнень як міра їх профілактики — антибіотикотерапія. Для хворих на муковісцидоз особливо небезпечна внутрішньогоспітальна інфекція, тому для них необхідні ізольовані приміщення (бокси) й деяку перевагу має амбулаторне лікування та кваліфіковане обслуговування в домашніх умовах, якщо вони відповідають усім вимогам, що встановлені лікарем.
ПЕРВИННІ ІМУНОДЕФІЦИТИ
Клінічні прояви (фенотип) кожної, особливо інфекційної та алергічної патології, залежить від імуногене-тичної програми індивідуума. Захисні імунні сили організму реалізуються в ході життєдіяльності 2 • 1012 клітин, роботи 4-х специфічних органів (тимусу, кісткового мозку, селезінки та лімфатичних вузлів).
В-лімфоцити у відповідь на зустріч організму з антигеном синтезують специфічні антитіла, імуногло-буліни G, М,А, Е та D ( залежно від будови важкого ланцюга молекули). Кожна молекула імуноглобуліну складається з чотирьох поліпептидних ланцюгів: 2 легких (має по одній варіабельній та константній частині) і 2 важких ( по одній варіабельній та три константні частини). Гени, які кодують імуноглобуліни, утворюють З групи зчеплення (дві для легких ланцюгів та одна для важкого ланцюга) на різних хромосомах. Узгоджена експресія цих генів забезпечує специфічність та своєчасність Імунних реакцій організму. В молекулі імуноглобуліну варіабельні ділянки (V) двох ланцюгів (легкого —L, важкого—Н) створюють «пастки для антигенів», акцепторні закінчення. Константні (С) ділянки двох важких (Н) ланцюгів утворюють ефекторний кінець молекули, від якого залежить частка специфічного комплексу антиген + антитіло: аглютинація, лізис, зв'язування комплементу та ін.
Схематичне зображення молекули гаммаглобуліну подано на рис. 12.
Т-клітини залежно від функції, яку вони виконують, можуть бути : помічниками (Т-хелпери), пригноблювачами (Т-супресори), вбивцями (Т-кілери). У цих клітинах працюють гени-модифікатори імунної відповіді, що кодують інтерферони, інтерлейкіни та інші імуномодулятори. Мутації в різних генах різних клітин зумовлюють весь спектр первинних імунодефіцитів. Під
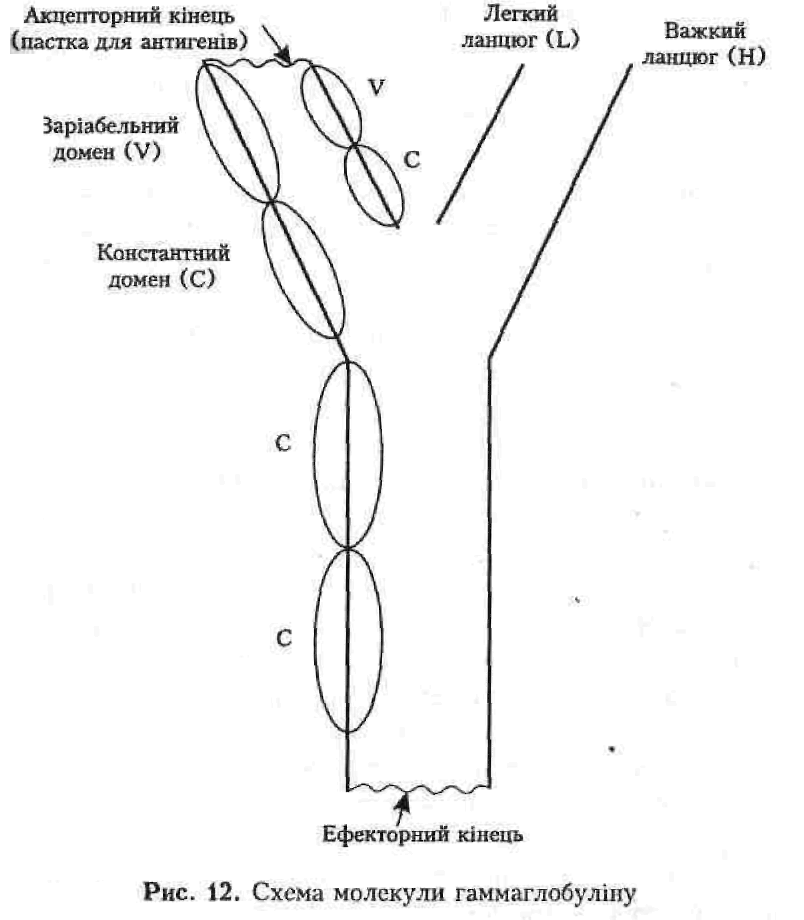
час кожної імунної відповіді створюється клон спеціалізованих лімфоцитів, які зберігають пам'ять про антиген. Щоразу під час створення імунітету клітини повинні інтенсивно розмножуватися, тому повинна багаторазово реплікуватися ДНК (проходити її синтез). Дефект одного з ферментів, що бере участь в синтезі ДНК — аденозиндезамінази (забезпечує гідролітичне дезамінування аденозину) є причиною тяжкої комбінованої імунної недостатності (ТКІН).
ТКШ проявляється від народження дитини лімфо-пенією й аплазією вилочкової залози, відсутністю гіпер-
чутливості уповільненого типу, порушенням синтезу імуноглобулінів. Лімфоцити хворих не реагують на мітогени in vitro. АР-тип успадкування. Етіологічне значення мають мутації в генах аденозиндезамінази та в гені активації рекомбінації (ГАР-1). Єдиним ефективним методом лікування в наш час вважається трансплантація гістосумісного кісткового мозку. До операції хворі діти потребують забезпечення стерильних умов існування (стерильні їжа, повітря, скафандр і т. ін.).
Серед найбільш розповсюджених спадкових імунодефіцитів слід зупинитися на таких.
Агаммаглобулінемія, гіпогаммаглобулінемія — проявляються у віці від б місяців до 2 років хронічними (пневмонії, синусити) інфекціями, що викликаються незвичними, іноді навіть непатогенними збудниками. При цьому спостерігається дефіцит імуноглобулінів G. Можливі поствакцинальні ускладнення. Існують форми з АР- та ХР- (хвороба Брутона) типом успадкування.
Селективний дефіцит імуноглобулінів А — звичайно починає проявлятися в 3 —7-річному віці. З цим імунодефіцитним станом пов'язані: ревматоїдний артрит, червоний вовчак, анкілозивний спондиліт, дерматоміозит, атонічний дерматит, ячміні, отит, лямбліоз, цукровий діабет, тиреоїдит та інші аутоімунні процеси. АР-тип успадкування, частина цих станів зчеплена з аномалією 18-ї хромосоми.
Зміна співвідношення імуноглобулінів М та G вбік збільшення концентрації IgM характерна для лімфа-денопатії.
При генетично зумовленому Ig Е-синдромі спостерігається непогана стійкість до звичайних інфекцій. Водночас у дітей поволі, майже непомітно з'являються холодні абсцеси, без болю та підвищення температури тіла, сверблячий дерматит, алергічні стани, кандидоз.
У дітей до 2 — 3 міс життя може спостерігатися тран-зиторна гіпогаммаглобулінемія, яка пов'язана з пізнім імунологічним стартом.
Імунопроліферативна хвороба — проявляється чутливістю до вірусу Епстейна-Барра та схильністю до захворювання на лімфому (навіть злоякісну) та інфекційний мононуклеоз. ХР-тип успадкування.
Гіпоплазія тимуса (синдром Ді Джорджі). З одного боку — це ПВР, з іншого — Т-клітинний імунодефіцит. Проявляється з народження судомами (гіпопара-тиреоз), підвищеною схильністю до вірусних та грибкових інфекцій, аномаліями розвитку кісткової системи, ротової порожнини, внутрішніх органів, аплазією або гіпоплазією вилочкової залози, недостатністю Т-клітин-ного імунітету. Без відповідного лікування смерть хворого настає до 2 років життя. Описані родини з АР-або АД-типом успадкування, делецією хромосоми 22q 11, спостерігаються спорадичні випадки.
Синдром Віскотта —Олдріча — імунодефіцит з тромбоцитопенією та екземою. Клінічно проявляється з народження: тромбоцитопенічна пурпура, рідкі випорожнення з домішками крові, згодом приєднуються пневмонія, отит, дерматити, екзема, можливе виникнення злоякісних пухлин, що нагадує ретикулоендотеліоз. Провідним є геморагічний синдром, генетичний'блок на рівні В-лімфоцитів. Тип успадкування — ХР.
Атаксія-телеангіектазія (синдром Луї —Бара) — комплексне захворювання імунної, нервової та ендокринної систем з частим ушкодженням шкіри та печінки. Одним з симптомів цієї патології є резистентний до інсуліну цукровий діабет, схильність до злоякісних пухлин ретикулярного типу. АР-тип успадкування.
Лікування імунодефіцитів, головним чином, симптоматичне: вітамінотерапія, антибіотики, гемотрансфузія; трансплантація кісткового мозку в наших умовах проблематична; імуномоделююча терапія може і буде використовуватися все більше в зв'язку з досягненнями генно-інженерної промисловості та зростаючими можливостями діагностики імунного статусу (дзеркала) кожного хворого.
СПАДКОВІСТЬ В ОНКОПАТОЛОГІЇ
Під час характеристики клінічного перебігу первинних імунодефіцитів ми згадували про зв'язок цієї генетичної патології з канцерогенезом. Пухлини — це насамперед молекулярна хвороба мутаційної природи. У генетичній програмі існують численні гени, що кодують або регулюють розмноження клітин, збільшення клітинної маси, диференціювання клітин, які створюють спеціалізовані тканини та органи. Найбільш напружено ці гени (онкогени та антионкогени) працюють протягом ембріонального розвитку і диференційовано, за складною системою регуляції їх експресії, виключаються з роботи при зміні генетичної програми після народження. У наш час вже відомо безліч онко-генів та антионкогенів (супресорних генів), які беруть, участь в контролі за проліферацією клітин та поділяються на 4 головні групи: 1) гени, що кодують розчинні фактори росту (група sis)', 2) гени, що кодують рецептори на поверхні клітин до фактора росту (групи Erb В, fms, neu)', 3) гени, продукти яких забезпечують передавання сигналу всередині клітин (групи, сімейства ras, src, abl)\ 4) гени ядерних білків, що беруть участь у ДНК-реплікації та транскрипції генів (тус, fos, myb, Rb, Wilms, p5^ та ін.).
Найчастіше ознака злоякісності клітини (втрата диференційованості, можливостей реагувати на сигнали інших клітин організму, неконтрольована проліферація) зумовлюється гомозиготним комплексом мутантних алелів, тобто наявністю двох мутацій в певному локусі гомологічних хромосом. Якщо обидві мутації виникають в соматичних клітинах організму, тоді злоякісна пухлина утворюється відносно рідко, на схилі життя, бо після випадкової мутації в онкогені (ініціація) проходить довгий період промоції, необхідний для знову ж таки випадкового виникнення другої мутації
в тому ж гені, але в гомологічній хромосомі. При цьому протягом періоду промоції можлива елімінація клітин з першою мутацією або репарація останньої і тоді пухлина не виникне. А якщо така злоякісна клітина і утвориться, то може не дати нащадків, оскільки імунна система організму не впізнає її як свою і знищить. Онко-генні віруси мають в своїй генетичній програмі вже готові мутантні онкогени і здатні вбудовувати їх у відповідні місця (замість або поряд з онкогенами клітин за механізмом комплементарності) одразу двох гомологів, утворюючи гомозиготу. Тоді онкогенез не потребує періоду промоції, і пухлини розвиваються скоріше (як при ВІЛ-інфекції). Цей вірус одночасно ушкоджує імунні клітини організму, викликає синдром набутого імунодефіциту (СНІД), для клінічної картини якого характерні злоякісні новоутворення (саркома Капоші, злоякісна лімфома, можливо, плоскоклітинна карцинома ротової порожнини та аденокарцинома прямої кишки у чоловіків-гомосексуалістів, інфікованих ВІЛ).
Не тільки набуті, але й успадковані імунодефіцитні стани, дефекти репаративної системи, хромосомні хвороби супроводжуються бластомогеннимй процесами.
Існує можливість отримання одного чи двох мутантних онкогенів у спадок від батьків при заплідненні. Тоді вже зникає необхідність в періодах ініціації, або Ініціації і промоції, пухлини з'являються в ембріональному або дитячому чи молодому віці. Усі клітини такого організму, що є клоном однієї клітини-зиготи, містять мутантний онкоген. Практично встановлено, що одна третина ембріональних пухлин — це спадкові форми.
Ретинобластома — злоякісне новоутворення, що походить з нервових клітин сітківки ока і виявляється у віці 1 — 1,5 років, хвороба проявляється своєрідним світінням зіниці на світло (котяче око),відмічається послаблена реакція зіниці на світло, косоокість, втрата зору, сліпота. Можливі метастази, ушкодження другого ока, кальци-фікація пухлини. Хвороба супроводжується розумовою
відсталістю. Описується АД-тип успадкування з пенет-рантністю більше як 90%. Ген ретинобластоми розташований на хромосомі 13ql4. При ретинобластомі мають місце мікроделеції в цьому локусі. Лікування: фо-толазерна коагуляція,енуклеація з подальшою хіміо- та рентгенотерапією. При своєчасному лікуванні можливе клінічне одужання.
Нефробластома — пухлина нирок, супроводжується вадами розвитку нирок та сечового міхура, аніридією. Ген, що кодує нефробластому, розташований в хромосомі Hql2-13, поряд з геном аніридії, каталази, лактатдегідрогенази, всіх ланцюгів гемоглобіну. АД-тип успадкування з пенетрантністю 63%, тобто 37% з усіх, хто має таку мутацію — носії без клінічних проявів хвороби. Можлива ДНК-діагностика онкогенетичних захворювань та носійства відповідних мутацій.
У тих випадках, коли всі клітини організму мають один мутантний онкоген, одержаний від зиготи, локалізація злоякісних пухлин буде залежати від того, в клітині якого органу виникла друга алельна мутація. Описані два типи сімейних раків: тип 1 — пухлини ендометрію, яєчників, молочної залози, простати, підшлункової залози, товстої кишки, шлунка, шкіри, легенів. Деякі з них входять до синдрому Li-Fraumeni, при якому відкриті заміни нуклеотидів в антионко-гені р53', тип 2 — рак молочної залози, саркоми, лейкози, пухлини мозку, рак сечового міхура.
Існує просто сімейний рак яєчників — менделююча патологія, мутація в гені erb B-2. Мутація в цьому ж гені описана при злоякісній пухлині товстої кишки. У випадках раку молочної залози виявлена мутація в онкогені На ras (білок р21, у 12-му кодоні заміна аденіну на гуанін).
Генетикам відомі спадкові синдроми множинних ендокринних неоплазій (МЕН): МЕН І (синдром Вер-нера), МЕН Па (синдром Сипла), МЕН 116 — синдром
гаевром слизових оболонок. Для цих генетичних менде-Ілюючих патологічних процесів характерними є пухлини щитоподібної, підшлункової, прищитоподібних залоз, гіпофіза та надниркових залоз. Вони мають АД-тип успадкування з дуже високою (майже 100%) пенетран-тністю. Ген, що зумовлює МЕН І, локалізований на хромосомі llq, МЕН II — на хромосомі 10q.
Зараз відомо більше як 200 спадкових синдромів, при яких існує схильність до розвитку злоякісних пухлин, серед них імунодефіцити, дефекти репаратив-них систем, факоматози (з групи генодерматозів).
Р.Ф.Гарькавцева (1992) виділяє такі молекулярні механізми онкогенезу: точкові мутації в протоонкоге-нах або антионкогенах; хромосомні перебудови з переміщенням протоонкогена під постійно діючий промотор; ампліфікація онкогена (при раку легенів у 34 рази); активація протоонкогена ретровірусною інсер-цією; активація протоонкогена, соматична мутація під впливом хімічних, фізичних та біологічних канцерогенів. Можливе порушення геномного імпринтингу.
На генодерматозах — генетично зумовлених захворюваннях шкіри, ми зупинятися не будемо, тому що вони добре викладені в спеціальній літературі. Це ж стосується і спадкових хвороб окремих органів та систем (нервової, серцево-судинної, кістково-м'язового апарату тощо).
МІТОХОНДРІАЛЬНІ ХВОРОБИ
Генетична програма організму записана послідовністю нуклеотидів в ДНК, що локалізується не тільки в хромосомах ядра, але й у цитоплазмі кожної клітини — в хромосомі мітохондрій. Код мітохондрій дещо відрізняється від того, що існує при записі в ДНК ядра, і був описаний у попередніх розділах. Гени, розташовані в ДНК мітохондрій, успадковуються не за законами Менделя, а шляхом прямого розподілу мітохондрій
між новими клітинами разом з цитоплазмою під час мітозу. Мітохондрії — це енергетичні системи клітин, ДНК яких називають М-хромосомою, або 25-ою хромосомою. Мітохондрії реп лікуються незалежно від хромосом ядра, вони4 присутні в цитоплазмі у великій кількості (ампліфіковані). Організм може бути гомо-хондріальним або гетерохондріальним залежно від того, однакові чи різні Мітохондрії існують у цитоплазмі його клітин.
Енергетичні потреби клітин забезпечуються за допомогою гліколізу чи окислювально-відновлювальних процесів. Останні здійснюються за рахунок роботи 60 генів (5 комплексів) дихального ланцюга мітохондрій, тільки 13 з яких локалізуються в М-хромосомі, інші — ядерні гени; між усіма генами існує тісний, добре скорегований взаємозв'язок. Генна карта М-хромосоми наведена на рис. 13.
Будь-яке порушення в генах дихального ланцюга супроводжується розвитком спадкової патології, а саме мітохондріальних хвороб. Мутації в генах, розташованих в ядерних хромосомах, е також причиною мітохондріальних хвороб, що успадковуються за законами Менделя. Якщо ушкоджені гени М-хромосом, — виникають хвороби з цитоплазматичною (материнською) успадкованістю, бо велику цитоплазму з мітохондрія-ми мають лише яйцеклітини. Тобто, в цьому випадку хворобу передають тільки жінки, але хворіють особи обох статей. Можливі спорадичні випадки мітохондріальних хвороб, нові мутації в ядерних, чи мітохондріальних генах. Ушкодження генів мітохондрій соматичних клітин може відбутися в окремих тканинах чи органах і викликати патологію з переважним ураженням певного органу.
Генетична класифікація мітохондріальних хвороб базується на їх розподілі по групах за умовами успадкування:
Дефекти ядерної ДНК, різні мутації, що зумовлю ють: а) дефекти транспорту субстрату; 6) дефекти утилізації субстрату; в) дефекти ферментів циклу Кребса; г) дефекти процесів окислювання-фосфори- лування; д) дефекти дихального ланцюга; е) дефекти постачання білків.
Дефекти мітохондріальної ДНК: а) спорадичні масштабні перебудови; б) точкові мутації в структур них генах, в) точкові мутації з генах, що взаємодіють зі структурними генами.
Дефекти міжгеномних сигналів: а) аутосомно-до мінантні множинні делеції мітохондріальної ДНК; б) аутосомно-рецесивне пригнічення функції мітохондрі альної ДНК.
Набуті пошкодження мітохондріальної ДНК під впливом дії шкідливих чинників: а) токсинів, наприк лад 1-метил-4-феніл-1,2,3,6-тетрагідропіридин; б) ліків, наприклад зидовудин (AZT) і в) процесу старіння.
До спорадичних випадків мітохондріальних хвороб, в основі яких лежать поодинокі делеції чи інсерції в деяких генах, належать синдром Кірнса —Сейра (оф-тальмоплегія, пігментна дегенерація сітківки, кардіомі-опатія, порушення піруват- та лактат-метаболізму, можлива інсерція вірусу); церебральна прогресуюча екстер-нальна офтальмоплегія (СРЕО); синдром Пірсона.
Серед мітохондріальних хвороб з материнським типом успадкування розрізняють синдроми: MERRF (міоклональна епілепсія, ураження м'язових волокон, точкова мутація в гені лізинової тРНК); MELAS (міто-хондріальна енцефаломіопатія з лактат-ацидозом та нападоподібним перебігом, точкова мутація в гені лей-цинової тРНК); міокардіопатія, атаксія-сліпота (точкова мутація в гені АТФ-ази 6); LHON (Лебера спадкова атрофія зорових нервів, декілька точкових мутацій в генах І комплексу дихального ланцюга (NADH dehydrogenase). Всі описані мутації змінюють гени, що розташовані в мітохондріях.
Менделюючі мітохондріальні хвороби представлені аутосомно-домінантною та аутосомно-рецесивною формами церебральної прогресуючої офтальмоплегії, рецидиву-ючою міоглобінурією (множинні делеції в генах, що працюють у клітинах м'язів) та нападами лактат-ацидозу (множинні делеції в генах, що працюють в клітинах м'язів та лімфоцитах). Поряд з цими якісними порушеннями спостерігається також тканеспецифічне зменшення кількості мітохондрій, що призводить до розвитку міопатії — нефропатії або гепатопатії, або енцефаломіопатії.
Таким чином, головними клінічними та біохімічними ознаками мітохондріальних хвороб є міопатії (енцефаломіопатії, кардіоміопатії), втрата зору за рахунок пігментного ретиніту, чи атрофії зорового нерва, порушення метаболізму пірувату та лактату, ферментів комплексів дихального ланцюга.
Діагностика мітохондріальних хвороб починається зі звичного для лікаря-генетика етапу складання родоводу та його аналізу для визначення типу успадкування патології в родині. Наступним — є клінічне обстеження хворого та членів його сім'ї, лабораторні аналізи стану метаболізму у хворого: визначення органічних кислот, амінокислот та карнітину у крові, у добовій сечі та цереброспінальній рідині (в залежності від клінічної ситуації). Результати цього обстеження визначають необхідність у наступному етапі досліджувати кров (прямий аналіз для визначення мутацій у мітохондріальній ДНК) чи матеріал біопсії м'язів: гістохімія та електронна мікроскопія (кількість та структура мітохондрій, їх функція), біохімічне дослідження дефектів окислювання-фосфорилування (у свіжих зразках біопсійного матеріалу) та аналіз ДНК мітохондрій.
Знання етіопатогенетичних механізмів мітохондріальних хвороб відкриває нові можливості їх специфічного лікування. Так, при лактат-ацидозі в залежності від первинного дефекту з успіхом використовуються різні підходи:
|
Дефект |
Терапія |
|
Глюкозо-6-фосфатаза |
Дієта, збагачена вуглеводами |
|
Фруктозо- 1 -6-дифосфатаза |
— »• |
|
Шруваткарбоксилаза |
— »• |
|
Фосфатдегідрогеназний комплекс |
Бідна на вуглеводи або кетогени дієта, діхлорацетат, тіамін, карнітин |
|
Біотинідаза |
Біотин |
|
Карбоксилаза |
Біотин |
|
NADH-CoQ редуктаза |
Нікотинамід, рибофлавін, карнітин |
|
Цитохромоксидаза |
Діхлорацетат, карнітин |
|
MELAS.MERRF, синдром Кірнса — Сейра |
Нікотинамід, рибофлавін, діхлорацетат, карнітин |
|
Дефекти окислювання жирних кислот |
Карнітин |
Аналіз мітохондріальної ДНК використовується також для встановлення спорідненості людей по материнській лінії. Так, з його допомогою були одержані вичерпні докази щодо царського походження розстріляних більшовиками в 1918 р. членів родини російського царя Миколи II шляхом порівняння ДНК мітохондрій клітин з ексгумованих останків і живих родичів царя за материнською лінією (рис. 14). Таким чином, вперше була продемонстрована наявність гете-ромітохондріальності та нової точкової мутації (заміна пар основ) у мітохондріальній ДНК жіночої яйцеклітини.
ХВОРОБИ ГЕНОМНОГО ІМПРИНТИНГУ
Цей клас хвороб належить до здобутків «нової генетики». Саме результати фундаментальних досліджень дозволили зрозуміти етіологію та патогенез багатьох спадкових хвороб і тих, що раніше до них не відносилися, як результат модифікації експресії генів, їх
хімічного змінений, яке не спричинює мутації, або структурного ушкодження самого гена. Тобто не змінюється текст інформації, але певні алелі виключаються, і надалі не функціонують. У цьому випадку ми маємо успадкування модифікацій. Клінічний перебіг хвороби, навіть наявність патології залежить від материнського чи батьківського походження алелів деяких генів. Імпринтинг означає маркерування на епігенетичному рівні, що відбувається під час гаметогенезу і викликає стійкі модифікації експресії гомологічних генів.
З відомих на сьогодні процесів, котрі повністю залежать від імпринтингу, треба зупинитися на ембріональному розвитку ссавців взагалі і людини зокрема. В останні роки вчені інтенсивно шукають відповіді на питання, чому ссавці є виключно залежними від статевої репродукції і як ця залежність відзначається на молекулярному рівні. Без відповідей на ці питання кло-нування ссавців не має наукової основи і не може бути успішно вирішеним. Імпринтинг — це чи не найбільша загадка нашого часу. Відомим є тільки один з його механізмів — метилювання цитозину в певних місцях ДНК. Приєднані до цитозину метилові групи виключають з експресії ген, і тоді працює тільки його неме-тильований алель, який може локалізуватися в материнському чи батьківському геномі. Тобто, імпринто-вана гемізиготність регулює ембріональний розвиток людини так само, як і інших ссавців. Такі, гени у мишей описані у вигляді кластерів на хромосомах 7, 17, X та інших, у людини — це кластери на хромосомах 11, 15, 6, X та інших. Під час ембріонального розвитку існують періоди моноалельної експресії генів, якщо вона порушується, виникають ушкодження плода або його оболонок. Гени, що пройшли чоловічий Імпринтинг (розташовані в батьківському геномі), кодують розвиток мембран плода та плаценти, а ті, що пройшли жіночий мейоз (в геномі яйцеклітини), керують розвитком тіла
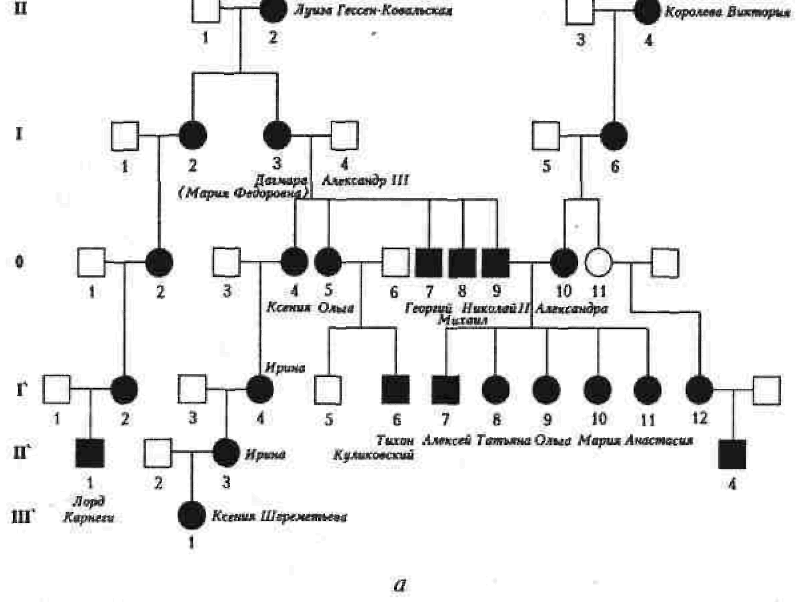
Рис. 14. а — Родовід царя Миколи II. За допомогою ДНК-
дактилоскопії Павлом Івановим із співавторами було
встановлено, що серед ексгумованих останків є члени родини: