

5 курс / Пульмонология и фтизиатрия / Чучалин_А_Г_Респираторная_медицина_т_1_2017
.pdf
Раздел
4 Респираторная патология и воспаление
4.1. Патология: рак легкого на фоне идиопатического легочного фиброза
Е.А. Коган, С.А. Демура
Идиопатический легочный фиброз (ИЛФ) с морфологией обычной интерстициальной пневмонии (ОИП) является одним из наиболее прогностически неблагоприятных заболеваний группы хронических ИЗЛ. Морфологический субстрат ОИП характеризуется наличием хронического воспаления, дисрегенераторными процессами и фиброзом легочного интерстиция респираторных отделов легкого, нарушением газообмена, прогрессирующей хронической ДН (ХДН), а также развитием рака легких [1, 5, 7, 31, 39]. Инициальным процессом в развитии рака легкого на фоне пневмосклероза является персистирующее повреждение легочной паренхимы, вызывающее хроническое воспаление с дисрегенераторным ответом, ведущее к злокачественной трансформации клеток. В этом разделе нами последовательно будут рассмотрены исторические аспекты данной проблемы, особенности персистирующего повреждения легочной паренхимы, репарации и этапы канцерогенеза рака на фоне ОИП.
История изучения вопроса о связи хронического воспаления, пневмосклероза и рака легкого
Проблема склероза и рака легкого не нова. История ее восходит к первым описаниям развития рака легкого в очагах фиброза, так называемого «рака легкого» в рубце. Впервые сообщение о раке легкого в рубце было сделано немецкими патологами Фридрих в 1939 г. и Росл в 1943 г., изучившими развитие рака легкого в посттуберкулезных рубцах и стенках хронических туберкулезных каверн [1, 6, 7, 12]. А.И. Струков описывал данные микроскопического исследования бронхогенного рака, растущего из бронха, замурованного в фи-
брозной ткани посттуберкулезного рубца [6]. По данным разных авторов, природа фиброзных изменений, на фоне которых возникает рак легкого, может быть различной, а не только туберкулезной: постинфарктной, при пневмокониозах, постпневмонической (очаги карнификации), вокруг инородных тел [7, 12, 17, 18, 25, 31].
Особый интерес в связи с изложенным выше представляет факт развития рака легкого на фоне ОИП при идиопатическом легочном фиброзе (ИЛФ), а также при пневмокониозах и системном склерозе. По данным М. Turner-Warwick и нашим наблюдениям, рак легкого на фоне ИЛФ развивается в 12,5–13% случаях [4, 5, 7, 39]. При этом в подавляющем числе случаев развивается аденокарцинома легких со стелющимся характером роста, которая в предыдущей классификации рака легких именовалась бронхиолоальвеолярным раком. Однако морфогенез склероза и дисрегенераторных изменений в заключительной стадии развития заболевания, «сотовом легком» остаются в настоящее время малоизученными.
Однако имеются и противники концепции развития рака легкого на фоне фиброза, утверждавшие, что фиброз является вторичной реакцией на рост злокачественной опухоли, называемой
десмопластической реакцией [25]. Данная точка зрения имеет право на существование, поскольку действительно рост опухоли сопровождается формированием десмопластической реакции на границе с сохранной тканью легкого. Впрочем, десмопластическая реакция и предсуществующий фиброз легкого — это не одно и то же, и между ними может быть проведена дифференциальная диагностика, что было проделано в последующих исследованиях [6–11]. Авторы опирались на анамнестические данные и динамические рентгенотомографические исследования, доказывающие наличие у больных раком легкого хронических заболеваний с прогрессирующим пневмосклерозом, морфологическую верификацию предрака и ранних стадий рака легкого в очагах пневмосклероза, когда еще сохранна структура предсуществующей легочной ткани, морфологические маркеры происхождения фиброзных изменений, состав коллагенов, обнаружение в фиброзных очагах предраковых изменений [7].
171

Раздел 4
Повреждение и хроническое воспаление в патогенезе рака легкого на фоне обычной интерстициальной пневмонии
Распространенность и персистенция повреждения при обычной интерстициальной пневмонии
Повреждение легочной паренхимы является инициальным процессом в развитии ОИП, при которой возникает сложный комплекс паренхима- тозно-стромальных взаимоотношений, одним из проявлений которых является развитие склероза в ответ на повреждение и в исходе хронического воспаления с дисрегенерацией. В.В. Серов и А.Б. Шехтер в монографии, посвященной патологии соединительной ткани, отмечают, что «… рост соединительной ткани ран кожи, дефектов различных тканей и органов или развитие склеротических процессов имеет общие закономерности. Этот процесс состоится из стадий повреждения, воспаления (включая микроциркуляторную, нейтрофильную и макрофагальную реакцию), пролиферации фибробластов и роста сосудов, вплоть до образования грануляционной ткани, контракции, ремоделирования (перестройки), инволюции или стабилизации фиброзной ткани» [20, 21].
Вопрос о природе повреждающего фактора при ОИП остается открытым. Обсуждается роль инфекционных агентов, среди которых особое место принадлежит вирусу Эпштейна–Барр, вирусу гепатита C, и острая респираторная вирусная инфекция (ОРВИ) [17, 18, 31]. Кроме того, лекарственные препараты, например цитостатики (метотрексат), антибиотики, противоаритмические препараты (амиодарон), противовирусные препараты, пищевые добавки, блокаторы кальциевых каналов также могут служить фактором повреждения легочного интерстиция [5, 31]. Существует также точка зрения о наличии генетической предрасположенности к развитию избыточного фиброзообразования в легких в ответ на неспецифическое повреждение эпителия [4, 5, 14–16]. Общим для всех перечисленных кандидатов в этиологические факторы ИЛФ является то, что они способны вызывать глубокое и персистирующее повреждение.
Вторым и, возможно, основным вариантом повреждения при ИЛФ является вторичное повреждение, вызванное агрессивными альвеолярными и ИМ, а также иммунопатологическими процессами (иммунокомплексными, антительными и цитотоксическими). Агрессивность альвеолярных и ИМ проявляется в том, что они генерируют активные формы кислорода, секретируют матриксные металлопротеиназы, фактор некроза опухолей и другие цитокины [6, 8, 17, 18, 31].
Резюмируя данные о повреждающих факторах при ОИП, следует отметить, что первичные и вторичные повреждающие факторы действуют парал-
лельно, постоянно вызывая тяжелые деструктивные изменения в респираторных отделах легких.
Особое значение, вероятно, имеет и локализация самого повреждения легочной паренхимы. Наиболее уязвимыми являются ниши стволовых клеток (НСК).
Локализация повреждения при обычной интерстициальной пневмонии
Немаловажным аспектом разбираемой проблемы является то, что при ОИП повреждение, а затем и последующее хроническое воспаления затрагивают наиболее уязвимую часть респираторных отделов легкого — нишу стволовых клеток (СК).
Согласно современным представлениям в ремоделировании легочной ткани участвуют СК как тканевого, так и костномозгового происхождения, прежде всего мезенхимальные стволовые костномозговые клетки. В тканях стволовые клетки располагаются в определенных структурных компартментах, называемых НСК, которые обеспечивают их жизнедеятельность в активном и дормантном состоянии, а также участие в процессах репарации [23, 35]. В респираторных отделах легких существует несколько зон, в которых формируются НСК и обнаруживаются стволовые клетки [24]. Такими зонами НСК респираторного ацинуса является бронхиолярно-альвеолярная переходно-клеточная зона (БАПЗ), где располагаются клетки Клара и интерстициальные клетки, зона стыка альвеол, где находятся пневмоциты 2-го порядка (Пн2п), и межальвеолярные капилляры [24]. Важная роль в осуществлении репаративных процессов помимо стволовых клеток принадлежит и прогениторным клеткам мезенхимального и эпителиального происхождения, перицитам и эндотелию сосудов, а также компонентам экстрацеллюлярного матрикса и базальным мембранам [24]. В связи с этим изучение повреждения зон НСК в патогенезе идиопатических интерстициальных пневмоний (ИИП) представляет особый интерес, поскольку эта гетерогенная группа заболеваний неуточненной этиологии характеризуется преобладанием диффузного и обычно хронического поражения интерстиция респираторных отделов легких, прежде всего альвеол и бронхиол [2, 3, 11].
По нашим данным, при ОИП патологические изменения респираторных отделов легких локализуются в БАПЗ и зоне стыка альвеол, где эпителиальная выстилка отсутствовала, что нередко сочеталось с разрушением базальной мембраны. Здесь же обнаруживается воспалительный инфильтрат с большим содержанием альвеолярных и ИМ (экспрессирующих CD68) с примесью полиморфноядерных лейкоцитов, а также лимфоцитов с образованием в легочной ткани «лимфоидных фолликулов». В просвете альвеол определяются полиморфноядерные лейкоциты. Участки разрушенной базальной мембраны замещаются SMA и виментин-позитивными миофибробластами [4, 5]
172

Респираторная патология и воспаление
и фибробластами [6], некоторые из которых экспрессировали CD117, Oct-4 и CD34 (рис. 4.1–4.4). Отмечается тромбоз микрососудов, сопровождаемый ишемией ацинарных отделов, что усиливало повреждение. При ОИП процесс распространялся на несколько зон НСК: БАПЗ, а также на места стыков альвеол, и вокруг микрососудов интерстиция [2–4].
В зонах НСК определяется не только усиление некроза эпителия с разрушением базальных мембран, но и повышение уровня апоптоза. При ОИП экспрессия Apo-Cas отчетливо была выражена в зонах НСК: в БАПЗ, в зонах стыка альвеол и вокруг микрососудов (капилляров и венул). Средний уровень экспрессии Apo-Cas в этих зонах в альвеолярном эпителии (АЭ) соста-
вил 0,7±0,2%, в эпителии аденоматозных структур (АдЭ) — 12,7±1,6%, в бронхиолярном эпителии — 8,2±1,6%, в АМ — 4,5±0,8% и миофибробластах (МФБ) — 2±0,6% [3, 4].
ОИП характеризовался достоверно более высокими уровнями экспрессии и других маркеров повреждения в отличие от контроля. Прямые корреляционные зависимости были обнаружены между уровнями экспрессии TGFβ в различных клетках зон НСК при ОИП: АЭ, бронхиолярный эпителий, МФБ, АМ и эндотелий. Уровень экспрессии TGFβ в АМ и МФБ напрямую коррелировал с уровнем экспрессии в этих клетках Apo-Cas.
Уровни экспрессии металлопротеаз были относительно высокими. MMP 7, отвечающая за реализацию эффектов TGFβ, экспрессировалась
Рис. 4.1. «Сотовое легкое» с лимфогистиоцитарной инфильтрацией, утолщением и склерозом альвеолярных перегородок и формированием сотовых (кистозных) структур. Окраска гематоксилином и эозином. ×200
Рис. 4.3. ИЛФ. То же наблюдение. Зона бронхиолоальвеолярной трансформации с пролиферацией миофибробластов и формированием миофибробластического фокуса, не окрашиваемого пикрофуксином. Окраска по Ван Гизону. ×400
Рис. 4.2. ИЛФ. Обычная интерстициальная пневмония. Зона бронхиолоальвеолярной трансформации с пролиферацией миофибробластов. Окраска гематоксилином и эозином. ×400
Рис. 4.4. ИЛФ. Обычная интерстициальная пневмония с пролиферацией миофибробластов и формированием миофибробластического фокуса. Гладкомышечный актин в миофибробластах миофибробластического фокуса зоны бронхиолоальвеолярной трансформации. Иммунопероксидазная реакция с выявлением гладкомышечного актина. ×400
173

Раздел 4
втех же зонах НСК, что и Apo-Cas, TGFβ и TNF-α. Уровень экспрессии MMP 7 коррелировал с экспрессией TGFβ в фибробласты, МФБ и эндотелий. Прямая корреляционная зависимость обнаружена между экспрессией MMP 7 и TNF-α
вАЭ, МФБ, фибробласты, АМ. MMP-2, протеаза, разрушающая мембраны, синтезирующаяся клетками воспалительного инфильтрата и в очагах неоангиогенеза при репарации, была обнаружена
вучастках разрушения базальных мембран в зонах НСК как в экстрацеллюлярном матриксе, так и
вАЭ, АдЭ, в бронхиолярном эпителии, в АМ, фибробласты, МФБ и эндотелий. Уровень экспрессии MMP 2 обратно коррелировал с уровнем экспрессии Apo-Cas и TNF-α в АМ, фибробласты, МФБ и эндотелий и коррелировал с экспрессией TGFβ и ММР 7 в АЭ, АМ, МФБ и фибробласты. Экспрессия MMP 1, «макрофагальной» протеазы, вызывающей повреждение респираторных отделов ацинуса, обнаруживалась при ОИП в АЭ, АдЭ, в бронхиолярном эпителии, АМ и МФБ. Уровни ее экспрессии в клетках зон ниш НСК коррелировали с уровнями экспрессии MMP 7, MMP 1, TNF-α и TGFβ.
Морфологическая картина ОИП, уровни экспрессии иммуногистохимических маркеров повреждения и локализация этой экспрессии свидетельствуют о глубоком повреждении зон НСК на всех уровнях респираторных отделов легкого, которое захватывает и БАПЗ, и зону стыка альвеол, и микрососуды, и сопровождается разрушением базальных мембран всего респираторного ацинуса, выраженным апоптозом эпителиоцитов и высоким уровнем экспрессии металлопротеаз. Такие клетки, как АМ, МФБ и фибробласты, вероятно, играют ключевую роль при развитии первичного и вторичного повреждения, судя по данным корреляционного анализа.
На ранней стадии ИЛФ обнаружены признаки повышенной готовности АЭ и бронхиолярного эпителия к апоптозу, судя по экспрессии ApoCAS, которая достоверно повышена в бронхиолоальвеолярной переходной зоне. В то же время экспрессии Apo-CAS в SMA-положительных МФБ не было зарегистрировано [11–13]. АЭ и бронхиолярный эпителий переходной зоны на ранней стадии ИЛФ подвергаются апоптозу, который приводит к патологической репарации с начинающимся формированием мелких очагов аденоматозной гиперплазии, экспрессирующей PCNA, PDGF. Пролиферирующие МФБ в миофибробластических фокусах переходной зоны экспрессируют PCNA, PDGF уже с ранней стадии заболевания. Статистически достоверная прямая корреляция между уровнем экспрессии PCNA миофибробластами и уровнем экспрессии АpoCAS альвеолоцитами в переходной зоне на ранней стадии свидетельствует о том, что миофибробластические фокусы, участвуя в апоптозе рядом лежащих альвеолоцитов, могут быть ключевым
фактором в прогрессировании фиброза легких с ранней стадией ИЛФ. С помощью подсчета сосудов с CD34-положительным эндотелием обнаружено достоверное повышение их плотности на ранней стадии ИЛФ по сравнению с контрольной группой нормальной ткани легких. Очаги неоангиогенеза локализуются преимущественно вблизи бронхиолоальвеолярной переходной зоне [11, 12, 14].
Хроническое воспаление при обычной интерстициальной пневмонии
Последние исследования вновь подтвердили роль хронического воспаления в патогенезе ОИП при ИЛФ [3, 6, 15, 27, 28, 31]. В очагах поражения обнаруживаются клеточные инфильтраты, продуцирующие разнообразные провоспалительные цитокины. Среди клеток воспалительных инфильтратов при ОИП встречаются в большом количестве альвеолярные и ИМ, нейтрофилы, эозинофилы, ТК. Считается, что центральной клеткой воспаления при альвеолитах является альвеолярный макрофаг. Макрофаги способны высвобождать хемоаттрактанты для нейтрофилов, включая лейкотриен В4 и IL-8, факторы роста (PDGF, инсулиноподобный фактор роста 1, TGFβ, фибронектин [15], стимулировать секреторную активность фибробластов и нейтрофилов [12], высвобождать кислородные радикалы, играющие важнейшую роль в повреждении паренхимы [12]. Нейтрофилы также являются основными эффекторными клетками при ИЛФ, способными высвобождать такие повреждающие факторы, как протеазы (коллагеназа, эластаза), кислородные радикалы [12]. Определенное значение имеют и Т-лимфоциты. У больных ИЛФ обнаруживают маркеры активации Т-клеток в крови, в сыворотке крови повышение IL-2 [12], а в жидкости БАЛ — IFN-γ [31]. IFN-γ активирует макрофаги и лимфоциты, стимулирует экспрессию эндотелиальных клеток молекул адгезии ICAM-1 и экспрессию продуктов HLA-DR, а также оказывает влияние на депозицию коллагена в интерстиции [12].
Одним из ключевых провоспалительных цитокинов при ОИП является TNF-α. Экспрессия TNF-α при ОИП обнаруживается в зонах в клетках воспалительных инфильтратов в зонах НСК. Наиболее высокий уровень TNF-α зафиксирован в макрофагах, МФБ и эндотелии сосудов [3].
Другим важным подтверждением существования хронического воспаления при ОИП является участие в прогрессировании заболевания TLR. TLR являются ключевыми молекулами врожденного иммунитета. Их основная функция сводится к формированию воспалительного ответа и отграничения внедрившегося повреждающего агента.
TLRs являются членами суперсемейства рецептора IL-1 (IL-1R), выполняющих ключевую роль в защите хозяина от инфекции. В легочной ткани они располагаются в различных клетках, в том числе Т-лимфоцитах, моноцитах, ДК, альвеолоци-
174

Респираторная патология и воспаление
тах II типа, эпителии бронхиол и бронхов, гладкомышечных клетках и фибробластах. TLR1, 2, 4, 5
и6 в основном экспрессируются на поверхности клеток и способны распознавать патоген-ассоции- рованные молекулярные паттерны бактерий, грибов и простейших. TLR3, 7, 8 и 9 располагаются в цитоплазме клеток и в первую очередь способны взаимодействовать с нуклеиновыми кислотами вирусов и бактерий. TLRs участвуют как в инициации врожденного иммунитета, так и в адаптивных иммунных реакциях. Активация TLR стимулирует антимикробный ответ путем усиления протеолиза микробных агентов, секреции сильнодействующих факторов, таких как дефенсинов и лизоцима, стимуляции оксидативного стресса и интенсификации фагоцитоза [15, 34]. В условиях декомпенсированной воспалительной реакции, склерозирования
ипри опухолевом росте активация TLR может способствовать прогрессированию этих процессов. Так показано, что при ИЛФ имеется усиление экспрессии TLR2 мРНК, TLR7 мРНК, TLR9 мРНК [31–34]. Другой аспект роли TLR в легочной ткани при патологических процессах связан с процессами репарации и опухолевым ростом.
При ИЛФ обнаружено достоверно более высокое содержание TLR2 мРНК, относящийся к экзосомальным рецепторам и активирующий продукцию провоспалительных и профиброгенных цитокинов, таких как NF-kB, ФНО- и IL-1β, что было показано на блеомициновой модели фиброза легких. Таким образом, TLR2 может быть новой мишенью для разработки терапевтических агентов против фибропролиферативных заболеваний легких [32, 34].
Активация эндосомальных TLR7 мРНК, TLR9 мРНК при ИФЛ также может способствовать хронизации воспаления и нарушению репарации в условиях повышенного апоптоза и гипоксических повреждений ткани легкого. При этом могут активироваться NF-kB, протеинкиназы, что ведет к запуску пролиферативных процессов в эпителиальных структурах, приводящих к развитию аденоматозных предраковых изменений [31, 34].
Резюмируя данные по TLR при ИФЛ следует отметить, что хронизация воспаления и развитие склероза, а также пролиферативных дисрегенераторных изменений эпителия в виде аденоматоза поддерживается и происходит при участии механизмов, запускаемых TLR 2, 7 и 9.
Заключение: обсуждая роль повреждения и хронического воспаления в прогрессировании ОИП и развитии рака легкого следует отметить, что ОИП отличается более глубоким повреждением НСК с развитием на их территории хронического воспаления с дисфункцией TLR 2, 7, 9 типов в клетках НСК на всех уровнях респираторных отделов легкого, которое распространяется и на БАПЗ, и на зону стыка альвеол, и на микрососуды и сопровождается разрушением базальных мембран всего респираторного ацинуса, выраженным апоптозом
эпителиоцитов и высоким уровнем экспрессии металлопротеаз.
Патологическая репарация и предрак и рак легкого на фоне интерстициального легочного фиброза
Патологическая репарация при ИЛФ обнаруживается на поздних стадиях развития заболевания, когда развивается перестройка легочной ткани как за счет склеротических изменений интерстиция, так и за счет нарушения дифференцировки и гиперплазии эпителия. При этом следует помнить общепатологическую закономерность, что репарация зависит от глубины повреждения [5, 7, 25, 31, 36]. Она различна в случаях интактных и поврежденных базальных мембран.
Повреждение с интактной базальной мембраной
Инфекционные агенты, шок и токсические формы кислорода вызывают повреждение АЭ, затем экссудацию в просвет альвеол:
а) рассасывание экссудата и пролиферацию пневмоцитов 2 с восстановлением структуры альвеол;
б) или организацией экссудата с образованием телец Масона (карнификацией).
Повреждение с разрушением базальной мембраны
Повреждение с разрушением базальной мембраны заканчивается фиброзом и рубцеванием. При этом альвеолярный макрофаг активирует синтез коллагена 1-го типа и протеогликанов клетками легочного интерстиция — фибробластами и МФБ (рис. 4.5, 4.6). Последние мигрируют в просвет альвеол и участвуют в формировании полей фиброза.
Поздняя стадия ИЛФ характеризуется типичными макроскопическими изменениями в виде уплотнения легочной ткани, которая при этом приобретает резиновую плотность, а также в виде понижения воздушности и эластичности с формированием ячеистых структур, напоминающих пчелиные соты, — «сотовое легкое».
При микроскопическом исследовании выявляются выраженный склероз интерстиция респираторных отделов легких и кистозная перестройка легочной ткани. Паренхима легкого замещается грубой соединительной тканью, в которую замурованы кистозно-расширенные воздухоносные пространства, структуры «сотового легкого», выстланные изнутри аденоматозно-гиперплазиро- ванным АЭ (см. рис. 4.3). Альвеолоциты I типа замещаются гиперплазированными альвеолоцитами II типа с признаками атипии и развитием очаговой аденоматозной гиперплазии и атипической
175

Раздел 4
Рис. 4.5. ИФЛ. Обычная интерстициальная пневмония. Рис. 4.6. Фибробласт стромы легкого. Электронограмма. Миофибробласты миофибробластического фокуса с пуч- ×18 000 ками миофиламентов и липидными каплями в цитоплазме.
Электронограмма. ×18 000
аденоматозной гиперплазии, которая встретилась в 75% случаях поздних стадий ОИП, а также с очагами плоскоклеточной метаплазии, дисплазией эпителия. Использование иммуногистохимических и электронно-микроскопического методов позволяют выявить тяжелую патологию, развивающуюся в области аэрогематического барьера, которая и проявляется в клинической картине в виде прогрессирующей ДН. Аэрогематический барьер блокируется и перестает функционировать как за счет выраженного фиброза интерстиция альвеолярных перегородок, так и за счет дисрегенераторных изменений в эпителиальной выстилке.
Легочный интерстиций расширяется за счет утолщения и редупликации эпителиальных и эндотелиальных базальных мембран, в нем происходит накопление всех типов коллагенов при резко
Рис. 4.7. ИФЛ. Обычная интерстициальная пневмония. Повышенное содержание коллагена 4-го типа в базальных мембранах альвеолярных капилляров и эпителиальных структур в «сотовом легком». Иммунопероксидазная реакция. ×200
увеличенном удельном весе труднодеградируемых коллагенов IV и V типов (рис. 4.7). В зону аэрогематического барьера внедряется большое количество коллагеновых волокон, активированных фибробластов, фиброцитов, клеток воспалительного инфильтрата, среди которых в эту стадию, особенно при предшествующем лечении кортикостероидами, преобладают лимфоидные элементы и гистиоциты. Фибробласты легочного интерстиция с признаками высокой синтетической активности выявляются не только в зоне аэрогематического барьера, между утолщенными базальными мембранами («интерпозиция»), но и в просветах альвеол и капилляров, что ведет к запустеванию микрососудов и развитию не только блока аэрогематического барьера, но и гипертензии в малом кругу кровообращения. У больных ОИП, по данным некоторых авторов, появляются особые клоны быстро пролиферирующих фибробластов. Организация же экссудата и белковой жидкости в просвете альвеол с последующей эпителизацией приводит к формированию телец Массона и карнификации.
При электронной микроскопии легочный эпителий на поздних стадиях ИЛФ подвергается перестройке. Альвеоциты I типа на больших участках замещаются альвеоцитами II типа с незрелыми осмиофильными мультиламеллярными тельцами, нередко с признаками незрелости и клеточного атипизма с экспрессией ЦК 7 и 19 (рис. 4.8–4.10). В связи с нарушенной продукцией сурфактанта и облитерацией бронхиол развиваются очаги ателектаза легочной ткани.
Дисрегенераторные изменения легочного эпителия при ФА могут стать предопухолевыми процессами и приводить к развитию рака легкого. Дисрегенераторные изменения при ОИП в нашем материале представлены аденоматозной гиперплазией АЭ и бронхиолярного эпителия в 32 случаях
176

Респираторная патология и воспаление
Рис. 4.8. ИФЛ. Обычная интерстициальная пневмония. Аденоматоз в «сотовом легком» с атипией эпителия. Окраска гематоксилином и эозином. ×200
Рис. 4.10. Аденокарцинома со стелющимся ростом с продукцией слизи на фоне обычной интерстициальной пневмонии (бронхиолоальвеолярный рак). Окраска по Крейбергу. ×200
Рис. 4.9. ИФЛ. Обычная интерстициальная пневмония. Аденоматоз в «сотовом легком» с атипией эпителия. Окраска гематоксилином и эозином. ×400
из 43 (74%); атипической аденоматозной гиперплазией в 27 из 43 случаев (62%); овальными и щелевидными структурами на фоне фиброза в 19 случаях из 43 (44%), плоскоклеточная метаплазия эпителия встречалась в 21% (7 из 43). По данным M. Turner-Warwik [26], рак легкого развивается у 12,5% больных идиопатическим ИЛФ.
В нашем материале рак легкого выявлен у 7 больных ИЛФ и системной склеродермией. Опухоли, как правило, имеют строение стелющейся аденокарциномы (бронхиолоальвеолярного рака), что может быть объяснено локализацией предопухолевых изменений при ОИП в респираторных отделах легких и развитием злокачественной трансформации клеток Клара и альвеолоцитов II (рис. 4.11).
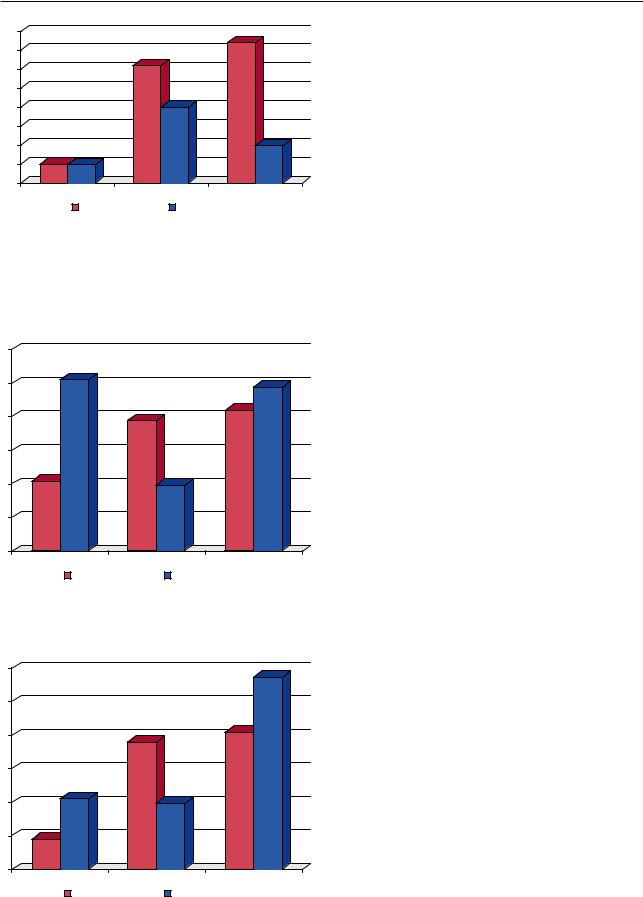
Уровень экспрессии TNF-α ИМ и фибробластами на поздней стадии ОИП снижается (см. рис. 4.11), а TGFβ нарастает (рис. 4.12). При этом средняя плотность капилляров в легочном интерстиции значительно уменьшилась. Уровень экспрессии TNF-α и FGFb клетками эпителия
альвеол и бронхиол был низок и составил соответственно 0,48±0,14 балла и 2,1±0,4 балла (рис. 4.13). Напротив, экспрессия TGFβ в эпителии относительно высокая и усиливалась на поздних стадиях (5,1±0,6 балла).
На поздней стадии ИЛФ также обнаружены признаки повышенной готовности АЭ и бронхиолярного эпителия в бронхиолоальвеолярной переходной зоне к апоптозу, судя по экспрессии Apo-CAS. В то же время в МФБ отмечен низкий уровень апоптоза. Данные свидетельствуют о том, что в процессе повреждения и репарации легочной ткани МФБ приобретают апоптоз-резистентный фенотип, способны к персистенции в ткани легких и становятся центральными клетками в прогрессировании фиброза легких у больных ИЛФ.
Статистически достоверная прямая корреляция между уровнем экспрессии PCNA миофибробластами и уровнем экспрессии Аpo-CAS альвеолоцитами в переходной зоне не только на ранней, но и на поздней стадии косвенно свидетельствует о том, что пролиферирующие МФБ миофибро-
177
Раздел 4 |
|
|
|
|
||
4 |
|
|
|
бластических фокусов участвуют в апоптозе ря- |
||
3,5 |
|
|
|
дом лежащих альвеолоцитов с ранней до поздней |
||
3 |
|
|
|
стадии ИЛФ. |
|
|
|
|
|
Поздняя стадия ИЛФ характеризовалась по- |
|||
|
|
|
|
|||
2,5 |
|
|
|
вышенными уровнями экспрессии PCNA, PDGF, |
||
2 |
|
|
|
EGFR во всех клетках легочной ткани по сравне- |
||
1,5 |
|
|
|
нию с ранней стадией заболевания. Данное повы- |
||
1 |
|
|
|
шение более заметно в клетках стромы переходной |
||
|
|
|
зоны со статистически достоверным повышением |
|||
0,5 |
|
|
|
|||
|
|
|
экспрессии PCNA, PDGF в МФБ на поздней ста- |
|||
|
|
|
|
|||
0 |
эпит |
АМ |
ИМ + фибр |
дии по сравнению с ранней стадией заболевания. |
||
|
На поздней стадии ИЛФ эпителиальные клетки |
|||||
|
Ранняя стадия |
Поздняя стадия |
||||
|
продуцируют PDGF, EGFR и тем самым усили- |
|||||
|
|
|
|
|||
Рис. 4.11. Экспрессия TNF-α при обычной интерстициаль- |
вают склеротические изменения. Интерстициаль- |
|||||
ной пневмонии (в баллах). Условные обозначения: эпит — |
ный склероз приводит к нарушениям регенерации |
|||||
клетки альвеолярного и бронхиолярного эпителия; АМ — |
эпителия в респираторных отделах легкого и мо- |
|||||
альвеолярные макрофаги; ИМ + фибр — интерстициаль- |
жет способствовать развитию дисплазии эпителия |
|||||
ные макрофаги и клетки фибробластического ряда интер- |
и рака легкого. |
|
||||
стиция респираторных отделов легких |
|
|||||
Проводимая нами оценка степени неоангио- |
||||||
|
|
|
|
|||
|
|
|
|
генеза доказала достоверное повышение плотно- |
||
|
|
|
|
сти новообразованных сосудов на поздней ста- |
||
6 |
|
|
|
дии ИЛФ по сравнению с контрольной группой |
||
|
|
|
|
нормальной ткани легких. Очаги неоангиоге- |
||
5 |
|
|
|
неза локализуются преимущественно в области |
||
|
|
|
|
бронхиолоальвеолярной переходной зоны рядом |
||
4 |
|
|
|
с аденоматозными структурами и вокруг очагов |
||
|
|
|
|
пневмосклероза. Хотя различие в плотности но- |
||
3 |
|
|
|
вообразованных сосудов на ранней и поздней ста- |
||
|
|
|
|
дии заболевания статистически не достоверно, но |
||
2 |
|
|
|
имеется тенденция к повышению интенсивности |
||
|
|
|
неоангиогенеза на поздней стадии по сравнению |
|||
|
|
|
|
|||
1 |
|
|
|
с ранней стадией заболевания. Наши данные еще |
||
|
|
|
раз подтверждают, что уровень ангиогенеза на бо- |
|||
|
|
|
|
лее поздней стадии ИЛФ не только не снижается, |
||
0 |
эпит |
АМ |
ИМ + фибр |
а наоборот, повышается. Очаги неоангиогенеза |
||
|
неравномерно располагаются в легочной ткани (в |
|||||
|
Ранняя стадия |
Поздняя стадия |
||||
|
основном вокруг очагов аденоматоза). |
|||||
|
|
|
|
|||
Рис. 4.12. Экспрессия TGFβ при обычной интерстициаль- |
Изучая роль MMPs и их тканевых ингибиторов |
|||||
ной пневмонии (в баллах) |
|
|
в процессе ремоделирования легочной ткани при |
|||
|
|
|
|
ИЛФ, мы обнаружили статистически достоверную |
||
|
|
|
|
прямую корреляцию между уровнями экспрессии |
||
6 |
|
|
|
MMP-7 и Apo-CAS гиперплазированными альве- |
||
|
|
|
олоцитами в очагах аденоматоза при ИЛФ как |
|||
|
|
|
|
|||
5 |
|
|
|
на ранней, так и на поздней стадии, что может |
||
|
|
|
свидетельствовать об участии MMP-7 в активации |
|||
|
|
|
|
апоптоза альвеолоцитов. |
|
|
4 |
|
|
|
Мы также установили, что на поздней стадии |
||
|
|
|
|
ИЛФ количество CD34-позитивных сосудов пря- |
||
3 |
|
|
|
мо коррелирует с уровнями экспрессии MMP-2 |
||
|
|
|
|
в фибро/миофибробластах, |
гиперплазированных |
|
2 |
|
|
|
альвеолоцитах, макрофагах |
и эндотелиоцитах. |
|
|
|
|
|
Наши результаты соответствуют данным литера- |
||
1 |
|
|
|
туры о роли MMPs в неоангиогенезе при ИЛФ. |
||
|
|
|
Доказанная нами высокая степень экспрессии |
|||
|
|
|
|
|||
0 |
|
|
|
MMPs при ИЛФ, особенно в БАПЗ, приводит к |
||
эпит |
АМ |
ИМ + фибр |
дисбалансу между MMPs и TIMPs. В результате |
|||
|
Ранняя стадия |
Поздняя стадия |
описанного процесса происходит не только бес- |
|||
|
|
|
|
контрольная деструкция легочной ткани в этой |
||
Рис. 4.13. Экспрессия FGFb при обычной интерстициаль- |
зоне, но и активация профибротических факто- |
|||||
ной пневмонии (в баллах) |
|
|
ров роста благодаря протеолитической активности |
|||
178

Респираторная патология и воспаление
MMPs, что способствует глубокой необратимой деструкции и нарушению архитектоники ткани легких.
Аденоматозная гиперплазия характеризуется пролиферацией разнообразных клеток: цилиндрических (клеток Клара), кубических (альвеоциты II типа), слизистых. В очагах аденоматозной гиперплазии обнаруживались ЦК 7-, ЦК 18- и ЦК 19-положительные клетки. ЦК 7- и ЦК 19-по- ложительные клетки встречались в основном в очагах пролиферирующих клеток (см. рис. 4.3).
При атипической аденоматозной гиперплазии
отмечаются увеличение размеров клеток, появление признаков клеточного и ядерного полиморфизма, в пролифератах появляются многоядерные и сосочковые структуры. Отмечается значительное увеличение ЦК 19-положительных клеток, тенденция к увеличению ЦК 7-положительных клеток, при этом количество ЦК 18-положитель- ных клеток остается на уровне аденоматозной гиперплазии.
Овальные и щелевидные структуры обнаруживались, как правило, в очагах грубых склеротических изменений и представляли собой «замурованные» в соединительной ткани бронхиолы и альвеолы, выстланные мономорфными кубическими клетками. Уровень экспрессии ЦК 7 в овальных и щелевидных структурах составил 62,4±9,8%, ЦК 19 — 79,7±4,5%, ЦК 18 — 35,2±3,7% (рис. 4.14).
Обобщая данные литературы и собственных исследований, можно выдвинуть гипотетическую схему морфогенеза ОИП, где ведущее значение играют клеточные кооперации, в центре которых стоят альвеолярные макрофаги, Т-лимфоциты, различные типы фибробластов, в ряде случаев —
нейтрофилы, эозинофилы, а также альвеолярный и бронхиолярный эпителий, включая эпителий переходной зоны.
Следует согласиться с мнением авторов, придерживающихся эпителиально-фибробластиче- ской концепции в патогенезе ИЛФ [36–38].
Эпителиально-фибробластический путь, по мнению Selman [36–38], характерен для ИЛФ и является ключевым моментом в развитии фиброзирования и аденоматозных изменений легочной паренхимы. Однако автор не учитывал того факта, что все клеточные переходы и трансформации происходят в нише стволовых клеток.
Так, по мнению Selman, потеря альвеолоцитарного защитного барьера приводит к тому, что базальная мембрана под действием кислорода разрушается (рис. 4.15). В ответе на разрушение базальной мембраны происходят регенерация и гиперплазия альвеолоцитов II типа. Гиперплазированные альвеолоциты II типа продуцируют различные факторы роста: ФНО-α, ФРК, TGFβ, инсулиноподобный фактор роста 1, ФРФ, привлекающие мезенхимальные клетки в очаги повреждения. Эти факторы роста стимулируют пролиферацию фибробластов, МФБ и продукцию компонентов межклеточного пространства [32]. Фибробласты, в свою очередь, способствуют повреждению альвеолоцитов путем индукции их апоптоза [29, 30].
Активированные интерстициальные и альвеолярные макрофаги вместе с нейтрофилами и другими клетками воспалительного инфильтрата выполняют важнейшую роль в повреждении эпителия, прежде всего переходной зоны, посредством генерации активных форм кислорода (АФК), протеаз, фактора некроза опухолей альфа, а также
Рис. 4.14. Уровень экспрессии ЦК 7, ЦК 18 и ЦК 19 клетками легочного эпителия в очагах дисрегенерации при обычной интерстициальной пневмонии (в процентах)
% |
|
|
|
|
100 |
|
|
|
|
90 |
|
|
|
|
80 |
|
|
|
|
70 |
|
|
|
|
60 |
|
|
|
|
50 |
|
|
|
|
40 |
|
|
|
|
30 |
|
|
|
|
20 |
|
|
|
|
10 |
|
|
|
|
0 |
|
|
|
|
АГ |
ААГ |
|
ОС |
ПМ |
|
ЦК 7 |
ЦК 18 |
ЦК 19 |
|
179