

Российская академия медицинских наук ГУ Российский онкологический научный центр им. Н.Н.Блохина
Научно-исследовательский институт канцерогенеза
КАНЦЕРОГЕНЕЗ
члена-корреспондента РАМН
Д.Г.Заридзе
РУКОВОДСТВО
Москва
"Медицина"
2004

ББК 55.6 УДК 616-006.6-092
К19
Канцерогенез / Под ред. Д. Г. Заридзе. — М.: Медицина, К19 2004. - 576 с: ил.: [2] л.ил. - ISBN 5-225-04787-4
Книга является руководством по фундаментальной онкологии. В ней представлены современные сведения, касающиеся эпидемиологии рака, химического, вирусного и радиационного канцерогенеза. Подробно рас смотрены генетические механизмы малигнизации клетки и влияние экзо генных факторов на процесс канцерогенеза. Главы, посвященные имму нологии рака, включают вопросы иммунологического контроля и отбора опухолевых клеток в процессе роста опухоли, а также современные дости жения в области иммунопрофилактики, иммунодиагностики и иммуноте рапии рака. Рассмотрены основные фундаментальные аспекты химио- и биотерапии опухолей. Описаны современные достижения в области кли нического применения результатов молекулярно-генетических исследова ний, в том числе и генной терапии.
Книга предназначена в качестве руководства по фундаментальным исследованиям в онкологии для широкого круга медицинских работников, биологов, студентов и аспирантов, работающих в этой области.
Carcinogenesis. Ed. by D. G. Zaridze. — Moscow: Meditsine, 2004. - 576 p.: ill. - ISBN 5-225-04787-4
The book is a manual of fundamental oncology. It presents the modern data on cancer epidemiology, chemical, viral and hormonal carcinogenesis. The man ual describes genetic mechanisms of cell malignization and influence of exoge nous and endogenous factors on the process of carcinogenesis. The chapters de voted to immunology of cancer, include questions on immunological control and selection of malignant cells during growth of tumor, and also modern achieve ment in areas immunoprevention, immunodiagnostics and immunotherapy. The
fundamental aspects |
of chemoand biotherapy of tumors are |
briefly considered. |
||
The modern achievements are described in the field of clinical application of re |
||||
sults of molecular genetics, including gene therapy. The book |
is intended as an |
|||
introductory manual |
of basic research |
in oncology for a wide |
range |
of physicians |
and biologists, including students and |
post-graduate students working |
in this area. |
||
|
|
|
|
ББК 55.6 |
ISBN 5-225-04787-4 |
|
© Коллектив авторов, 2004 |
||
Все права авторов защищены. Ни одна часть этого издания не может быть занесе на в память компьютера либо воспроизведена любым способом без предварительного письменного разрешения издателя.
Посвящается памяти
Леона Манусовича Шабада
( 1 9 0 2 — 1 9 8 2 ) ,
академика АМН СССР,
выдающегося ученого, основателя отечественной школы экспериментальной онкологии
Авторский коллектив
АБЕЛЕВ Г. И. — доктор биологических наук, профессор, заслуженный деятель науки РФ, академик РАН, руководитель лаборатории иммунохимии, профессор МГУ (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
АЛЬТШТЕЙН А. Д. — доктор биологических наук, профессор, заслуженный деятель науки РФ, академик РАЕН, руководитель лаборатории генетики виру сов (Институт биологии гена РАН).
БЕЛИЦКИЙ Г. А. — доктор медицинских наук, профессор, руководитель лабо ратории методов скрининга канцерогенов (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
БЕРШТЕЙН Л. М. — доктор медицинских наук, профессор, руководитель ла боратории онкоэндокринологии (НИИ онкологии им. Н. Н. Петрова МЗ РФ).
ВАСИЛЬЕВ Ю. М. — доктор медицинских наук, профессор, руководитель ла боратории механизмов канцерогенеза, член-корреспондент РАН, профессор МГУ (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
ГУРЦЕВИЧ В. Э. — доктор медицинских наук, профессор, руководитель лабо ратории вирусного канцерогенеза (НИИ канцерогенеза РОНЦ им. Н. Н. Бло хина РАМН).
ДЕЙЧМАН Г. И. — доктор медицинских наук, профессор, руководитель лабо ратории противоопухолевого иммунитета, заслуженный деятель науки РФ (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
ЕГОРОВ Е. Е. — кандидат биологических наук, ведущий научный сотрудник (Институт молекулярной биологии им. В. А. Энгельгардта РАН).
ЗАРИДЗЕ Д. Г. — доктор медицинских наук, профессор, заслуженный деятель науки, руководитель отдела эпидемиологии и профилактики опухолей, дирек тор НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН, член-корреспондент РАМН.
ЗБОРОВСКАЯ И. Б. — кандидат биологических наук, ведущий научный со трудник (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
ИЛЬИН К. в. — доктор медицинских наук.
КАРАМЫШЕВА А. Ф. — кандидат биологических наук, ведущий научный со трудник (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
КИСЕЛЕВ Ф. Л. — доктор биологических наук, профессор, руководитель лабо ратории молекулярной биологии вирусов (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
КИСЕЛЕВА Н. П. — кандидат биологических наук, ведущий научный сотруд ник (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
КЛЕЙМАН А. М. — кандидат медицинских наук.
КОБЛЯКОВ В. А. — доктор биологических наук, профессор, главный научный сотрудник (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
КОПНИН Б. П. — доктор медицинских наук, профессор, руководитель лабора тории цитогенетики (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
КРЮКОВА И. Н. — доктор биологических наук, заслуженный работник здра воохранения РФ (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
КУДРЯВЦЕВ И. А. — кандидат медицинских наук, ведущий научный сотруд ник (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
КУПРАШ Д. В. — кандидат биологических наук, ведущий научный сотрудник (Институт молекулярной биологии им. В. А. Энгельгардта РАН).
ЛАВРИК И. Н. — кандидат химических наук, ведущий научный сотрудник (Институт молекулярной биологии им. В. А. Энгельгардта РАН).
ЛИХТЕНШТЕЙН А. В. — доктор биологических наук, профессор, руководи тель лаборатории биохимии опухолей (НИИ канцерогенеза РОНЦ им. Н. Н. Бло хина РАМН).
МОРОЗОВ В. А. — доктор медицинских наук.
МЯСИЩЕВА Н. В. — доктор медицинских наук, профессор, заслуженный ра ботник здравоохранения РФ, руководитель лаборатории эндогенных модифи цирующих факторов (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
НЕДОСПАСОВ С. А. — доктор биологических наук, профессор, член-коррес пондент РАН, руководитель лаборатории (Институт молекулярной биологии им. В. А. Энгельгардта РАН).
ПЫЛЕВ Л. Н. — доктор медицинских наук, профессор, руководитель лабора тории природных канцерогенов (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
РОВЕНСКИЙ Ю. А. — доктор медицинских наук.
СЕНЮТА Н. Б. — кандидат медицинских наук, ведущий научный сотрудник (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
СТАВРОВСКАЯ А. А. — доктор медицинских наук, профессор, руководитель лаборатории генетики опухолевых клеток (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
ТАТОСЯН А. Г. — доктор медицинских наук, профессор, руководитель лабора тории регуляции клеточных и вирусных канцерогенов (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
ТУРУСОВ В. С. — доктор медицинских наук, профессор, руководитель лабора тории канцерогенных веществ (НИИ канцерогенеза РОНЦ им. Н. Н. Блохина РАМН).
ФЛЕЙШМАН Е. В. — доктор медицинских наук, профессор (НИИ канцероге неза РОНЦ им. Н. Н. Блохина РАМН).
Оглавление
Принятые сокращения и термины |
|
15 |
|||
Введение. — Д. Г. Заридзе |
|
25 |
|||
Г л а в а 1. |
Эпидемиология и этиология злокачественных новообразований. |
— |
|
||
|
|
Д. Г. Заридзе |
|
29 |
|
1.1. |
Методологические подходы к изучению этиологии опухолей человека . . . . |
29 |
|||
1.2. |
Дескриптивная эпидемиология |
|
33 |
||
1.3. |
Основные факторы риска злокачественных опухолей и профилактика . . . . |
44 |
|||
|
1.3.1. |
Курение и другие формы потребления табака |
|
44 |
|
|
1.3.2. |
Питание |
|
|
48 |
|
1.3.3. |
Эндогенные и экзогенные гормоны |
|
54 |
|
|
1.3.4. |
Потребление алкогольных напитков |
|
59 |
|
|
1.3.5. |
Профессиональные канцерогены |
|
61 |
|
|
1.3.6. |
Загрязнение воздуха |
|
63 |
|
|
1.3.7. |
Ультрафиолетовое излучение |
|
64 |
|
|
1.3.8. |
Ионизирующая радиация |
|
66 |
|
|
1.3.9. |
Инфекционные факторы |
|
73 |
|
|
1.3.10. Наследственность |
|
82 |
||
Рекомендуемая литература |
|
85 |
|||
Г л а в а 2. |
Основные свойства неопластической клетки и базовые механизмы их воз |
||||
|
|
никновения. — Б. П. Копнш |
86 |
||
2.1. |
Характерные признаки опухолевой клетки |
|
86 |
||
2.2. |
Механизмы возникновения характерных свойств неопластической клетки |
|
91 |
||
|
2.2.1. |
Нарушения регуляции клеточного цикла |
|
91 |
|
|
2.2.2. |
Изменения морфологии и движения клеток |
|
94 |
|
|
2.2.3. |
Отсутствие репликативного старения (иммортализация) |
|
94 |
|
|
2.2.4. |
Изменения регуляции апоптоза |
|
97 |
|
|
2.2.5. |
Генетическая нестабильность |
|
100 |
|
Рекомендуемая литература |
|
102 |
|||
Г л а в а 3. |
Молекулярно-генетические изменения в злокачественных клетках . . . . |
103 |
|||
3.1. |
Онкогены. — А. Г. Татосян |
|
103 |
||
|
3.1.1. |
Как идентифицируются онкогены? |
|
103 |
|
|
3.1.2. |
Механизмы активации протоонкогенов |
|
111 |
|
|
3.1.3. |
Онкогены в системе передачи сигналов |
|
114 |
|
|
|
3.1.3.1. |
Сигналы, стимулированные P D G F |
|
115 |
|
|
3.1.3.2. |
Тирозинкиназы в системе передачи сигналов |
|
118 |
|
|
3.1.3.3. |
G-белки в передаче внутриклеточных сигналов |
|
120 |
3.1.3.4."Ядерные" протоонкогены и факторы транскрипции в системе
|
передачи сигналов |
121 |
3.1.3.5. |
Протоонкогены — регуляторы гибели клетки |
122 |
Рекомендуемая литература |
124 |
|
3.2. Опухолевые супрессоры и мутаторные гены. — Б. П. Копнин |
125 |
|
3.2.1. pRb — первый идентифицированный опухолевый супрессор |
127 |
|
3.2.1.1. |
Открытие гена Rb |
127 |
3.2.1.2. Функция pRb в клетке и ее нарушения при канцерогенезе |
. . 128 |
|
3.2.1.3. |
Гомологи pRb: р 107 и Rb2/p 130 |
130 |
3.2.2.р53 — многофункциональный опухолевый супрессор, чаще всего пора
жаемый в различных новообразованиях человека |
130 |
3.2.2.1. Типы опухолей, ассоциированные с аномалиями р53 |
130 |
6

3.2.2.2.Структурная организация и биохимические активности белка
р53 |
132 |
3.2.2.3.Физиологические функции р53 и их нарушения в неопластиче
ских клетках |
135 |
3.2.2.4. Гомологи р53: рбЗ и р73 141
3.2.3.Продукты гена INK4a — pl6INK4a и pARF, регулирующие активность pRb
и р 5 3 |
142 |
3.2.4.Опухолевый супрессор PTEN регулирует клеточный цикл и апоптоз,
модулируя сигнальный путь PI3K-PKB/Akt |
144 |
3.2.5.Е-кадхерин, АРС и аксин — супрессоры, контролирующие сигнальный
путь (З-катенин/Cdk/pRb |
145 |
3.2.6. Компоненты сигнальных путей TGFp-Smad |
147 |
3.2.7.Опухолевый супрессор VHL регулирует реакцию на гипоксию и инги-
бирует ангиогенез |
148 |
3.2.8.TV/7/нейрофибромин и ЛТО/мерлин — гены нейрофиброматоза . . . . 148
3.2.9. |
Ген WT1 и опухоль Вильмса |
149 |
3.2.10. |
Мутаторные гены |
150 |
3.2.10.1. ATM, ATR, NBS1, СНК1 и С Н К 2 - компоненты систем про ведения сигналов от поврежденной Д Н К к различным эффек
торам |
|
150 |
3.2.10.2. BRCA1 |
и BRCA2 контролируют репарацию Д Н К и |
размноже |
ние клеток |
152 |
|
3.2.10.3. MSH2, |
MSH6, MLH1 и PMS2 — компоненты систем репара |
|
ции неспаренных оснований Д Н К |
154 |
|
3.2.10.4.Компоненты системы эксцизионной репарации Д Н К и пиг
ментная ксеродерма |
155 |
Рекомендуемая литература . , |
156 |
3.3.Цитокины, их рецепторы и передача внутриклеточных сигналов. — С. А. Не-
доспасов, Д. В. Купраш |
158 |
|
3.3.1. |
Классификация цитокинов и их рецепторов |
158 |
3.3.2. |
Некоторые важнейшие цитокины |
165 |
3.3.3. |
Цитокины и опухолевые клетки |
165 |
3.3.4. |
Цитокины и противоопухолевый надзор |
166 |
3.3.5.Цитокины в противоопухолевой терапии. Применение цитокинов in vit
ro |
166 |
3.3.6. Противоопухолевое применение цитокинов in vivo |
167 |
Рекомендуемая литература |
168 |
3.4.Механизмы активации программируемой клеточной гибели через рецепторы
смерти. — С. А. Недоспасов, И. Н. Лаврик |
168 |
|
3.4.1. |
Рецепторы смерти |
169 |
3.4.2. |
Цитокины, активирующие рецепторы смерти |
170 |
3.4.3. |
Каспазы |
171 |
3.4.4.Адапторные молекулы рецепторов смерти и каскад сигналов, активи
рующих каспазы |
173 |
3.4.5.Образование многокомпонентного белкового комплекса (DISC), запус
|
кающего механизм клеточной гибели |
175 |
3.4.6. |
Механизмы защиты клеток от инструктивного апоптоза |
176 |
3.4.7. |
Роль инструктивного апоптоза in vivo |
176 |
3.4.8.Роль инструктивного апоптоза при иммунном ответе на опухолевые
клетки и в канцерогенезе |
176 |
3.4.9.Перспективы направленного использования инструктивного апоптоза в
. противоопухолевой терапии |
178 |
|
Рекомендуемая литература |
179 |
|
3.5. Роль теломеразы в канцерогенезе. — Е. Е. Егоров |
179 |
|
3.5.1. История открытия теломеразы |
179 |
|
3.5.1.1. |
Смертные и бессмертные клетки |
179 |
3.5.1.2. |
Открытие теломеразы |
180 |
3.5.1.3. |
Теломерный повтор |
181 |
3.5.1.4. |
Тонкая структура теломер |
182 |
7

3.5.1.5. |
Теломеризация клеток |
183 |
3.5.1.6. |
Границы применимости теории недорепликации |
183 |
3.5.2. Теломераза и опухоли |
184 |
|
3.5.2.1. |
Роль репрессии теломеразы Б соматических клетках |
184 |
3.5.2.2. |
Процесс иммортализации клеток человека |
185 |
3.5.2.3.Каким образом опухолевые клетки преодолевают репрессию
|
теломеразы? |
186 |
3.5.2.4. |
Нетеломеразное удлинение теломер у человека |
187 |
3.5.3. Теломераза, теломеры и лечение опухолей |
187 |
|
3.5.3.1. |
Значение теломеразы в диагностике опухолей |
187 |
3.5.3.2. |
Подавление теломеразы как метод лечения опухолей |
188 |
3.5.3.3.Возможные способы воздействия на опухоль, опосредованные
|
теломеразой и теломерами |
. ) . . . |
189 |
|
3.5.3.3.1. Вакцинация или генерация цитотоксических Т-лим- |
|
|
|
фоцитов |
|
189 |
|
3.5.3.3.2. Ингибиторы обратных транскриптаз |
|
189 |
|
3.5.3.3.3. Олигонуклеотидные стратегии |
|
189 |
|
3.5.3.3.4. Воздействие на экспрессию генов теломеразы |
. . . . |
190 |
|
3.5.3.3.5. Воздействие на теломеры |
|
190 |
Рекомендуемая литература |
|
191 |
|
3.6. Эпигенетические изменения в опухолевых клетках. Роль метилирования Д Н К |
|
||
в канцерогенезе. — Н. П. Киселева, А. В. Лихтенштейн |
|
191 |
|
3.6.1. |
Роль метилирования Д Н К в нормальных клетках |
|
192 |
3.6.2. |
Роль деметилирования Д Н К в нормальной клетке |
|
196 |
3.6.3. |
Нарушения метилирования Д Н К при канцерогенезе |
|
197 |
3.6.4. |
Гипометилирование Д Н К при канцерогенезе |
|
201 |
Рекомендуемая литература |
|
202 |
|
Г л а в а 4. |
Химический канцерогенез |
204 |
|
4.1.Механизмы действия и классификация химических канцерогенов. — В. С. Ту
русов, Г. А. Белицкий, Л. Н. Пылев, В. А. Кобляков |
204 |
4.1.1. Генотоксические канцерогены |
204 |
4.1.1.1.Метаболическая активация и детоксикация химических канце
|
рогенов |
206 |
4.1.1.2. |
Полициклические ароматические углеводороды |
209 |
4.1.1.3. |
Нитрозосоединения |
211 |
4.1.1.4. |
Ароматические амины |
211 |
4.1.1.5. |
Афлатоксины |
212 |
4.1.2. Негенотоксические канцерогены |
212 |
|
4.1.2.1.Особенности канцерогенного эффекта негенотоксических кан
церогенов |
213 |
4.1.2.2.Механизмы действия негенотоксических канцерогенов . . . . 214
4.1.2.3.Классификация негенотоксических канцерогенов по механиз
му их действия |
215 |
4.1.2.3.1. Гормоны |
215 |
4.1.2.3.2. Соединения с эстрогеноподобным действием (ксено- |
|
гормоны) |
215 |
4.1.2.3.3. Тиреотропные канцерогены |
216 |
4.1.2.3.4.Стимуляторы пролиферации пероксисом (СПП) . . . 216
4.1.2.3.5.Цитотоксичные соединения, вызывающие длительную
клеточную пролиферацию |
217 |
4.1.2.3.6. Ингибиторы или стимуляторы ферментных систем, |
|
участвующих в регуляции передачи митотического |
|
сигнала |
217 |
4.1.2.3.7. Соединения, стимулирующие образование активных |
|
форм кислорода |
217 |
4.1.2.3.8. Прочие негенотоксические соединения |
218 |
4.1.3. Соединения с неустановленным механизмом действия |
218 |
4.1.3.1. Металлы |
218 |
8

4.1.3.2. |
Природные волокнистые и неволокнистые силикаты |
219 |
4.1.4. Трансплацентарный и трансгенерационный канцерогенез |
220 |
|
4.1.4.1. |
Трансплацентарный канцерогенез |
220 |
4.1.4.2. |
Трансгенерационный (мультигенерационный) канцерогенез |
220 |
4.1.5.Классификации химических канцерогенов по степени канцерогенной опасности для человека (так называемые гигиенические классифика
ции) |
221 |
Рекомендуемая литература |
225 |
4.2.Выявление и мониторинг химических канцерогенов. — Г. А. Белицкий, В. С. Ту
русов |
|
|
225 |
|
4.2.1. |
Скрининг канцерогенов по химическому строению |
|
226 |
|
4.2.2. |
Биологические методы скрининга |
|
227 |
|
|
4.2.2.1. |
Хронические эксперименты на животных |
|
227 |
|
|
4.2.2.1.1. Перспективы совершенствования |
|
229 |
|
|
4.2.2.1.2. Среднесрочные испытания in vivo — выявление орга- |
|
|
|
|
нотропности канцерогенов |
|
229 |
|
|
4.2.2.1.2.1. Печеночная модель |
|
229 |
|
|
4.2.2.1.2.2. Мультиорганные модели |
|
230 |
|
|
4.2.2.1.2.3. Модели опухолей отдельных органов . . . |
230 |
|
|
|
4.2.2.1.3. Трансплацентарный и неонатальный канцерогенез |
231 |
|
|
|
4.2.2.1.4. Нокаутные и трансгенные животные |
|
. 2 3 1 |
|
4.2.2.2. |
Скрининг канцерогенов в краткосрочных тестах (КСТ) . . . . |
234 |
|
|
|
4.2.2.2.1. Мутагенез и канцерогенез |
|
235 |
|
|
4.2.2.2.2. Метаболическая активация проканцерогенов в систе |
||
|
|
ме скрининговых тестов |
|
236 |
|
|
4.2.2.2.3. Принципы формирования батарей КСТ |
|
237 |
|
|
4.2.2.2.3.1. Минимальная батарея КСТ |
|
238 |
|
|
4.2.2.2.4. Индивидуальные скрининговые тесты |
|
238 |
|
|
4.2.2.2.4.1. Определение мутагенности с помощью бак |
||
|
|
териальных тест-систем |
|
238 |
|
|
4.2.2.2.4.2. Определение мутагенности на дрозофиле |
240 |
|
|
|
4.2.2.2.4.3. Индукция прямых генных мутаций в куль |
||
|
|
тивируемых клетках грызунов |
|
241 |
|
|
4.2.2.2.4.4. Индукция хромосомных аберраций в клет |
||
|
|
ках костного мозга мышей и микроядер |
||
|
|
ный тест |
|
241 |
|
|
4.2.2.2.4.5. Учет ДНК-повреждающего действия |
. . . |
241 |
|
4.2.2.3. |
Прямые ускоренные методы |
|
242 |
|
|
4.2.2.3.1. Индукция опухолей у аквариумных рыб |
|
242 |
|
|
4.2.2.3.2. Неопластическая трансформация клеток грызунов в |
||
|
|
культуре ткани |
|
242 |
|
4.2.2.4. |
Тесты для скрининга опухолевых промоторов |
|
243 |
4.2.2.5.Правила продвижения исследуемых веществ по батарее К С Т и
|
|
интерпретация результатов испытаний |
243 |
4.2.3. |
Мониторинг канцерогенов |
245 |
|
|
4.2.3.1. |
Физико-химический мониторинг |
245 |
|
4.2.3.2. |
Биологический мониторинг |
245 |
|
|
4.2.3.2.1. Мониторинг отдельных генотоксических |
канцероге |
|
|
нов человека . . |
246 |
|
|
4.2.3.2.2. Чувствительность биомаркерного показателя |
247 |
|
|
4.2.3.2.3. Репрезентативность тканей, используемых |
для целей |
|
|
мониторинга |
248 |
|
|
4.2.3.2.4. Индивидуализация мониторинга действия канцероге |
|
|
|
нов |
248 |
Рекомендуемая литература |
250 |
||
Г л а в а 5. |
Вирусный канцерогенез и роль вирусов в возникновении опухолей челове |
||
|
ка. — А. Д. Апътштейн |
251 |
|
5.1. Общая характеристика онкогенных вирусов |
251 |
||
9
5.1.1. |
Онкогенность вирусов |
253 |
5.1.2. |
Трансформирующая активность вирусов в культуре клеток |
255 |
5.1.3.Особенности взаимодействия онкогенных вирусов с клетками, транс
|
формированными ими in vitro или in vivo |
257 |
|
5.1.4. |
Онкогенные вирусы и онкогенные инфекции |
260 |
|
5.1.5. |
Полиома- и папилломавирусы (papovaviridae) |
261 |
|
|
5.1.5.1. |
Полиомавирусы |
262 |
|
5.1.5.2. |
Папилломавирусы |
263 |
5.1.6. |
Онкогенные аденовирусы |
263 |
|
5.1.7. |
Онкогенные герпесвирусы |
266 |
|
5.1.8. |
Онкогенные поксвирусы |
266 |
|
5.1.9. |
Онкогенные гепаднавирусы |
267 |
|
5.1.10. Ретровирусы (Retroviridae) |
268 |
||
|
5.1.10.1. |
Экзогенные и эндогенные ретровирусы |
271 |
5.1.10.2.Ретровирусы — возбудители опухолевых заболеваний . . . . . 272
Рекомендуемая литература |
274 |
5.2.Онкогенный потенциал вирусов и механизмы его проявления. — Ф. Л. Киселев 274
5.2.1. Трансформация клеток вирусами in vitro . . |
275 |
5.2.2.Вирусиндуцированная трансформация как модель для изучения контро
ля клеточного цикла и пути передачи сигнала |
276 |
5.2.3.Механизмы трансформации клеток РНК-содержащими вирусами . . . 277
5.2.4.Онкогены как компоненты регуляторной системы клетки, контроли
|
рующей пролиферацию и дифференцировку |
279 |
|
5.2.5. |
Дмс-активация |
под действием вирусов |
280 |
5.2.6. |
Онкогенность, |
индуцируемая /и^днс-активирующими ретровирусами |
282 |
5.2.7.Механизм трансформации клеток ДНК-содержащими вирусами . . . . 282
|
5.2.8. |
Онкогены ДНК-содержащих вирусов |
283 |
|
|
|
5.2.8.1. |
Аденовирусы |
284 |
|
|
5.2.8.2. |
Вирусы группы полиомы |
284 |
|
|
5.2.8.3. |
Вирусы папиллом |
285 |
|
5.2.9. |
Общие механизмы действия онкобелков ДНК-содержащих вирусов |
285 |
|
|
|
5.2.9.1. |
Инактивация функций белка гена ретинобластомы |
285 |
|
|
5.2.9.2. |
Инактивация функций р53 |
286 |
Рекомендуемая литература |
287 |
|||
5.3. |
Вирусы папиллом и их роль в канцерогенезе шейки матки. — Ф. Л. Киселев |
287 |
||
|
5.3.1. Участие вирусов папиллом в канцерогенезе шейки матки |
287 |
||
|
5.3.2. |
Регуляция транскрипции вирусных генов |
289 |
|
|
5.3.3. |
Трансформирующие гены вирусов папиллом |
290 |
|
|
5.3.4. Функциональная кооперация онкобелков Е6 и Е7 |
295 |
||
Рекомендуемая литература |
297 |
|||
5.4. |
Роль вируса гепатита в развитии рака печени. — Ф. Л. Киселев |
297 |
||
|
5.4.1. |
Геном вируса гепатита В |
298 |
|
|
5.4.2. |
Белок НВх |
299 |
|
|
5.4.3. |
Рге82-активаторы: LHBs и MHBs(t) |
300 |
|
|
5.4.4. |
Роль хронической инфекции HBV для развития опухолей печени . . . |
301 |
|
|
5.4.5. |
Геном вируса гепатита С |
302 |
|
Рекомендуемая литература |
303 |
|||
5.5. ДНК-содержащие вирусы: герпесвирусы. — В. Э. Гурцевич |
303 |
|||
|
5.5.1. |
Вирус Эпштейна—Барр. Общая характеристика |
305 |
|
|
5.5.2. |
Молекулярно-генетическая организация |
306 |
|
|
5.5.3. |
Продукты вирусных генов |
307 |
|
|
|
5.5.3.1. |
Экспрессия генов латентной инфекции |
307 |
|
|
5.5.3.2. |
Гены продуктивного цикла инфекции |
311 |
5.5.4.Дифференциальная экспрессия генов EBV в латентно инфицированных
клетках |
312 |
5.5.5.Экспрессия вирусных генов при EBV-ассоциированных заболеваниях
человека |
313 |
Рекомендуемая литература |
314 |
5.6. Вирус герпеса человека 8-го типа (HHV-8). — В. Э. Гурцевич |
314 |
10 |
|
>

5.6.1. |
Морфология и молекулярно-генетическая организация |
315 |
|
5.6.2. |
Функции вирусных генов, контролирующих пролиферацию клеток |
317 |
|
5.6.3. |
Биологические свойства вируса. Клеточный тропизм |
320 |
|
5.6.4. |
Эпидемиология HHV-8 |
321 |
|
|
5.6.4.1. |
Данные PCR-анализа |
^321 |
|
5.6.4.2. |
Данные серологических исследований |
322 |
|
5.6.4.3. |
Пути передачи инфекции |
323 |
5.6.5. |
Заболевания человека, связанные с HHV-8 |
323 |
|
|
5.6.5.1. |
Саркома Капоши |
323 |
|
5.6.5.2. |
Лимфопролиферативные заболевания |
324 |
Рекомендуемая литература |
325 |
||
5.7. Ретровирусы: вирус Т-клеточного лейкоза человека. — В. Э. Гурцевич |
325 |
||
5.7.1. |
Вирус Т-клеточного лейкоза человека . . |
326 |
|
5.7.1.1.Структура генома HTLV-I и основные кодируемые белки . . . 327
|
5.7.1.2. |
Основные свойства Tax-белка HTLV-I |
328 |
|
5.7.1.3. |
Географическое распространение HTLV-I инфекции |
329 |
|
5.7.1.4. |
Пути передачи инфекции |
329 |
|
5.7.1.5. |
Заболевания человека, ассоциированные с HTLV-I |
329 |
|
5.7.1.6. |
Роль различных факторов для HTLV-I-ассоциированного |
кан |
|
|
церогенеза |
331 |
5.7.2. Происхождение и распространение HTLV-I на планете |
332 |
||
Рекомендуемая литература |
333 |
||
5.8. Ретровирусы типа D (SRV). — В. А. Морозов, К. В. Ильин |
334 |
||
5.8.1. |
Морфогенез SRV |
335 |
|
5.8.2. |
Геном SRV |
336 |
|
5.8.3. |
Рецепторы и нейтрализационные эпитопы SRV |
337 |
|
5.8.4.Особенности заболевания обезьян, вызванного ретровирусами группы
|
SRV |
337 |
|
5.8.5. |
Экспериментальная передача заболевания |
|
337 |
5.8.6. Распространение вируса в организме обезьян |
|
338 |
|
5.8.7. |
Эпидемиология иммунодефицита обезьян |
|
338 |
5.8.8. SRV, выделенные из стабильных клеточных линий человека |
|
339 |
|
5.8.9. |
Распространение SRV среди людей |
|
339 |
5.8.10. Ретровирусы типа D овец и коз |
|
340 |
|
Рекомендуемая литература |
|
342 |
|
5.9. Эндогенные ретровирусы человека. — Н. Б. Сенюта, А. М. Клейман |
|
342 |
|
5.9.1. |
Происхождение эндогенных ретровирусов человека |
|
343 |
5.9.2. Строение и обнаружение эндогенных ретровирусов |
|
344 |
|
5.9.3. |
Классификация эндогенных ретровирусов |
|
345 |
5.9.4. |
Биологическая роль эндогенных ретровирусов |
|
345 |
Рекомендуемая литература |
|
351 |
|
5.10. О возможном участии ретровирусов в индукции рака молочных желез челове |
|
||
ка. — И. Н. Крюкова |
351 |
|
|
5.10.1. Краткие сведения о MMTV |
|
352 |
|
5.10.2. Сравнение канцерогенеза в молочной железе мыши и человека |
. . . . |
354 |
|
5.10.3. Попытки прямого обнаружения вируса в Р М Ж человека |
|
354 |
|
5.10.4. Поиски антител в сыворотках больных РМЖ, распознающих структур |
|
||
|
ные антигены MMTV |
|
355 |
5.10.5. Антигены, родственные структурным белкам MMTV у человека |
. . . . |
356 |
|
5.10.6. Последовательности Д Н К , гомологичные MMTV в геноме человека |
356 |
||
5.10.7. Экспрессия последовательностей Д Н К , гомологичных гену env |
MMTV, |
|
|
|
в лимфоцитах периферической крови человека |
|
359 |
Рекомендуемая литература |
|
361 |
|
Г л а в а 6. |
Гормональный канцерогенез. — Л. М. Берштейн, В. С. Турусов |
362 |
|
6.1. Механизмы гормонального канцерогенеза (ГК) |
|
362 |
|
6.1.1. |
Канцерогенез, вызываемый эстрогенами |
|
362 |
6.1.2. |
Канцерогенез, вызываемый неэстрогенами |
|
365 |
11

6.2.Генетический полиморфизм ферментов стероидогенеза, рецепторов стероид
|
ных гормонов и гормональный канцерогенез |
|
367 |
|
6.3. Гормональный канцерогенез, связанный с пре- и перинатальным развитием |
|
368 |
||
6.4. Трансплацентарный гормональный канцерогенез и беременность |
|
369 |
||
6.5. |
Трансгенерационный гормональный канцерогенез |
|
370 |
|
6.6. |
Гормональный канцерогенез и старение |
|
370 |
|
6.7. |
Гормоны и химический канцерогенез |
|
372 |
|
6.8. |
Ксеноэстрогены и эстрогенсодержащие лекарственные препараты |
|
372 |
|
6.9. |
Тканевая зависимость гормонального канцерогенеза у человека |
|
374 |
|
Рекомендуемая литература |
|
375 |
||
Г л а в а 7. |
Морфогенетические реакции клеток и их нарушения при опухолевой |
|
||
|
|
трансформации. — Ю. А. Ровенский, Ю. М. Васильев |
|
376 |
7.1. |
Цитоскелет . . . . : |
|
376 |
|
|
7.1.1. |
Актиновые микрофиламенты |
. |
. 376 |
|
7.1.2. |
Микротрубочки |
|
379 |
|
7.1.3. |
Промежуточные филаменты |
|
381 |
7.2. |
Распластывание и локомоция нормальных клеток |
|
382 |
|
|
7.2.1. |
Внеклеточный матрикс |
|
382 |
|
7.2.2. |
Морфология процесса распластывания клеток |
|
383 |
|
7.2.3. Фокальные контакты: структура, динамика и функции |
|
385 |
|
|
7.2.4. |
Локомоция клеток |
|
388 |
7.2.5.Роль малых ГТФаз в регуляции актинового цитоскелета, формировании
псевдоподий и локомоции клеток |
390 |
7.2.6. Биомеханический контроль морфогенеза и функций клеток |
394 |
7.2.7.Реакции клеток на химическую анизотропию и топографию внеклеточ
ного матрикса |
396 |
7.3.Нарушения распластывания и локомоции клеток в результате неопластической
трансформации |
399 |
|
7.3.1. |
Морфологические изменения |
399 |
7.3.2. |
Нарушения структуры и функций фокальных контактов |
401 |
7.3.3. |
Изменения локомоторных реакций клеток |
403 |
7.3.4. |
Изменения топографических реакций клеток |
405 |
7.4.Межклеточные контактные взаимодействия и" их нарушения при неопластиче
ской трансформации |
406 |
7.4.1. Контактные взаимодействия нормальных клеток |
406 |
7.4.2.Нарушения межклеточных контактных взаимодействий и их роль в опу
холевой инвазии |
409 |
Рекомендуемая литература |
414 |
Г л а в а 8. Эндогенные модуляторы канцерогенеза. — Н. В. Мясшцева, И. А. Кудрявцев |
415 |
8.1.Модулирующее влияние гликозаминогликанов и протеогликанов в процессах
опухолевого роста |
415 |
8.1.1.Модулирующее влияние гликозаминогликанов и протеогликанов на ге-
мопоэз |
415 |
8.1.1.1. Роль гепарансульфата в гемопоэзе |
. . . 416 |
8.1.1.2.Синтез и секреция ГАГ кроветворными клетками при лейкозах 417
8.1.1.3. |
Модулирующее влияние ГАГ на митогенную активность b F G F |
418 |
8.1.2. Экспрессия ГАГ и ПГ опухолевыми клетками |
418 |
|
8.1.2.1. |
Экспрессия ГС-ПГ при опухолях легких человека |
419 |
8.1.2.2. |
Экспрессия СД-1 при раке желудка |
419 |
8.1.2.3. Экспрессия СД-44 в опухолевых клетках |
419 |
|
8.1.2.4. |
Модулирующее влияние ХС-ПГ при раке простаты |
420 |
8.2.Метаболиты каскада арахидоновой кислоты как активные модуляторы канце
рогенеза |
421 |
8.2.1. Биосинтез и общие свойства эйкозаноидов . . . |
421 |
8.2.2.Роль каскада арахидоновой кислоты в инициации и промоции опухо
лей, индуцированных химическими канцерогенами |
423 |

8.2.3.Взаимодействие эйкозаноидов с онкогенами и другими эндогенными
факторами |
424 |
8.2.4. Роль метаболитов арахидоновой кислоты в иммуносупрессии |
424 |
8.2.5. Роль различных эйкозаноидов в контроле роста опухолевых клеток |
425 |
8.2.6.Влияние эйкозаноидов на дифференцировку опухолевых клеток . . . . 426
8.2.7.Роль различных метаболитов арахидоновой кислоты в метастазировании
|
|
опухолевых клеток |
|
424 |
||
Рекомендуемая литература |
|
427 ^ |
||||
Г л а в а 9. |
Ангиогенез опухоли: механизмы, новые подходы к терапии. — А. Ф. Кара- |
|
||||
|
|
мышева |
|
|
429 |
|
9.1. |
Ростовые факторы клеток эндотелия сосудов |
|
431 |
|||
9.2. |
Рецепторы V E G F |
|
|
435 |
||
9.3. Семейство рецепторов Tie и взаимодействующие с ними ростовые факторы |
|
439 |
||||
9.4. |
Ингибиторы ангиогенеза |
|
444 |
|||
Рекомендуемая литература |
|
447 |
||||
Г л а в а 10. |
Опухолевые |
антигены и противоопухолевый иммунитет (врожденный и |
|
|||
|
|
приобретенный). — Г. И. Дейчман |
448 |
|
||
10.1. Антигены опухолевых клеток |
|
451 |
||||
10.2. Специфические трансплантационные опухолевые антигены |
|
452 |
||||
10.3. Эффекторные механизмы противоопухолевого иммунитета |
|
458 |
||||
10.4. Иммунологический надзор организма и отбор опухолевых клеток в процессе |
|
|||||
|
роста и прогрессии опухоли |
|
468 |
|||
Рекомендуемая литература |
|
473 |
||||
Г л а в а 11. Иммунология опухолей человека. — Г. И. Абелев |
474 |
|
||||
11.1. Иммунодиагностика |
|
474 |
||||
11.2. Иммунопрофилактика |
|
477 |
||||
11.3. Иммунотерапия |
|
|
|
479 |
||
Рекомендуемая литература |
|
481 |
||||
Г л а в а 12. Практические выходы молекулярной генетики |
483 |
|
||||
12.1. Практические выходы современной цитогенетики опухолей. — Е. В. Флейишан |
483 |
|||||
|
12.1.1. Основные цитогенетические термины |
|
484 |
|||
|
12.1.2. Основные методические приемы |
|
484 |
|||
|
12.1.3. Клиническое значение хромосомного анализа в онкологии |
|
486 |
|||
|
|
12.1.3.1. Изменения кариотипа и малигнизация (краткие сведения) |
. . |
486 |
||
|
|
12.1.3.2. Диагностика и дифференциальная диагностика опухолей . |
. . |
494 |
||
|
|
12.1.3.3. |
Прогностическое значение хромосомного анализа |
|
501 |
|
|
12.1.4. Хромосомный анализ и мониторинг опухолей |
|
508 |
|||
Рекомендуемая литература |
|
512 |
||||
12.2. Молекулярно-биологические исследования онкогенов и генов-супрессоров |
|
|||||
|
опухолевого роста в клинической практике. — И. Б. Зборовская |
|
513 |
|||
|
12.2.1. Наследуемые |
особенности структуры онкогенов и генов-супрессоров |
|
|||
|
|
как факторы риска развития онкопатологии |
|
515 |
||
|
|
12.2.1.1. |
Мутации генов |
|
515 |
|
|
|
12.2.1.2. |
Полиморфизм онкогенов |
|
515 |
|
|
12.2.2. Досимптоматическая диагностика опухолей с использованием молеку- |
|
||||
|
|
лярно-генетического тестирования онкогенов и антионкогенов . . . . |
518 |
|||
|
|
12.2.2.1. |
Мутации генов |
|
518 |
|
|
|
12.2.2.2. |
Специфические делеции |
|
520 |
|
|
|
12.2.2.3. |
Экспрессия генов |
|
521 |
|
|
12.2.3. Молекулярно-генетические исследования в дифференциальной диагно |
|
||||
|
|
стике |
|
|
|
522 |
|
|
12.2.3.1. Гены регуляции клеточного цикла |
|
522 |
||
|
|
12.2.3.2. |
Экспрессия онкогенов и генов-супрессоров как фактор диффе- |
|
||
|
|
|
ренцировки клеток |
|
523 |
|
|
|
12.2.3.3. |
Мутации генов |
|
524 |
|
13

12.2.4. Генетические альтерации как факторы прогнозирования течения опухо |
|
|||||
левого процесса |
|
|
|
|
524 |
|
12.2.4.1. Полиморфизм онкогенов |
|
|
527 |
|||
12.2.4.2. |
Мутации генов |
|
|
|
527 |
|
12.2.4.3. Специфические делеции |
|
|
528 |
|||
12.2.4.4. Амплификация генов и увеличение их экспрессии как факторы |
|
|||||
|
прогноза |
|
|
|
530 |
|
12.2.5. Генетические маркеры и тактика лечения |
|
|
532 |
|||
12.2.6. Основные методы экспериментального анализа структурных и функ |
|
|||||
циональных изменений генов |
|
|
|
535 |
||
Рекомендуемая литература |
|
|
|
|
538 |
|
12.3. Генная терапия на основе вирусных векторов. — В. А. Морозов |
|
539 |
||||
12.3.1. ДНК-содержащие вирусы |
|
|
|
540 |
||
12.3.1.1. Аденовирусы |
|
|
|
540 |
||
12.3.1.2. Аденоассоциированные вирусы |
|
|
541 |
|||
12.3.1.3. |
Вирус простого герпеса 1 человека |
|
|
541 |
||
12.3.2. Невирусные способы доставки |
|
|
|
542 |
||
12.3.3. РНК-содержащие вирусы |
|
|
|
542 |
||
12.3.3.1. Принципы создания ретровирусного вектора на основе вируса |
|
|||||
|
лейкоза мышей Mo-MuLV |
|
|
543 |
||
12.3.3.2. |
Необходимые элементы ретровирусных векторов |
|
544 |
|||
12.3.3.3. |
Структура некоторых ретровирусных конструкций |
|
547 |
|||
12.3.3.4. |
Клетки для упаковки |
|
|
|
548 |
|
12.3.4. Использование методов генетической терапии в клинике . |
|
550 |
||||
12.3.4.1. Генетические болезни |
|
|
|
550 |
||
12.3.4.2. |
Инфекционные заболевания |
|
|
551 |
||
12.3.4.3. |
Онкологические заболевания |
|
|
551 |
||
12.3.4.4. |
Заболевания Ц Н С и опухоли мозга |
|
|
553 |
||
12.3.5. Перспективы генетической терапии |
|
|
554 |
|||
12.3.5.1. Векторы, находящиеся в разработке |
|
|
554 |
|||
Рекомендуемая литература |
|
|
|
|
557 |
|
Г л а в а 13. Механизмы лекарственной устойчивости опухолевых клеток. — А. А. Став- |
|
|||||
ровская |
|
|
|
|
558 |
|
13.1. Лекарственная устойчивость, обусловленная снижением накопления препарата |
|
|||||
внутри клетки |
|
|
|
|
|
560 |
13.1.1. Множественная |
лекарственная |
устойчивость, |
обусловленная Р-глико- |
|
||
протеином (Pgp-МЛУ) |
|
|
|
560 |
||
13.1.2. Множественная лекарственная устойчивость, определяемая белками се |
|
|||||
мейства M R P |
|
|
|
|
565 |
|
13.1.3. Множественная лекарственная устойчивость и белок ABCG2 (MXR/ |
|
|||||
BCRP) |
|
|
|
|
|
566 |
13.1.4. Множественная |
лекарственная |
устойчивость, |
определяемая |
белком |
|
|
LRP/Mvp |
|
|
|
|
567 |
|
13.2. Лекарственная устойчивость, обусловленная обезвреживанием препарата в |
|
|||||
клетке |
|
|
|
|
|
567 |
13.3. Лекарственная устойчивость, связанная с изменением или повышенной репа |
|
|||||
рацией мишени препаратов |
|
|
|
569 |
||
13.4. Роль ключевых генов, контролирующих апоптоз, в лекарственной устойчиво |
|
|||||
сти опухолевых клеток |
|
|
|
|
569 |
|
13.4.1. Ген р53 и лекарственная устойчивость опухолевых клеток |
|
569 |
||||
13.4.2. Антионкоген PTEN и лекарственная устойчивость опухолевых клеток |
571 |
|||||
13.4.3. Влияние онкогена BCL-2 и других генов семейства BCL-2 на лекарст |
|
|||||
венную устойчивость опухолевых клеток |
|
|
571 |
|||
13.4.4. Система CD95-L/CD95 (FasL/Fas) и лекарственная устойчивость |
. . . |
572 |
||||
13.4.5. Фактор транскрипции NF-KB |
|
|
|
572 |
||
Рекомендуемая литература |
|
|
|
|
574 |
|
14
Принятые сокращения и термины
ААВ |
— аденоассоциированные вирусы |
|
|
|
|||
ААФ (2-ААФ) |
— гидрокси-2-ацетиламинофлюорен |
|
|
|
|||
АГ |
— |
аппарат Гольджи |
|
|
|
|
|
АДА |
— |
аденозиндезаминаза |
|
|
|
|
|
АК |
— |
арахидоновая кислота |
|
|
|
||
ак, аа |
— аминокислота, amino acid |
|
|
|
|||
АКТГ |
— адренокортикотропный гормон |
|
|
|
|||
А П К |
— |
антигенпрезентирующие клетки |
|
|
|
||
Апоптоз |
— программированная гибель клеток |
|
|
||||
АР |
— атрибутивный риск, т. е. процент всех случаев рака, |
этиологи |
|||||
|
|
чески связанный с курением |
|
|
|
||
АФП |
— альфа-фетопротеин, |
сывороточный белок, |
продуцируемый |
||||
|
|
клетками желточного мешка и печени и являющийся серологи |
|||||
|
|
ческим маркером соответствующих опухолей |
|
|
|||
БаП, БП |
— бенз(а)пирен |
|
|
|
|
|
|
ББД |
— белок Бенс-Джонса, |
моноклональные Ig L-цепи, поступающие |
|||||
|
|
из крови в мочу |
|
|
|
|
|
Б К |
— бластный криз хронического миелолейкоза |
|
|
||||
БОЕ |
— бурстобразующая единица |
|
|
|
|||
БХ (HD) |
— болезнь Ходжкина, |
лимфогранулематоз |
|
|
|||
БХМЭ |
— бисхлорметиловый эфир |
|
|
|
|||
ВИЧ-1 (HIV) |
— вирус иммунодефицита человека |
|
|
|
|||
ВОЗ |
— Всемирная организация здравоохранения |
|
|
||||
ВПГ-1 |
— вирус простого герпеса 1 |
|
|
|
|||
ВРМЖ |
— вирус рака молочной железы мышей |
|
|
||||
ВЭБ |
— вирус Эпштейна—Барр |
|
|
|
|||
ГАГ |
— |
гликозаминогликаны |
|
|
|
||
ГАУ |
— гиперпластический |
альвеолярный |
узелок |
|
|
||
ГВБ |
— главный внутренний белок |
|
|
|
|||
ГГТ |
— |
у-глютамилтранспептидаза |
|
|
|
||
ГГФРТ |
— |
гипоксантингуанинфосфорибозилтрансфераза |
|
||||
ГДФ |
— |
гуанозиндифосфат |
|
|
|
|
|
Гемобластоз |
— опухоль кроветворной системы |
|
|
|
|||
Гены "домашнего хозяйства" |
|
|
|
|
|
||
("housekeeping" гены) |
— конститутивно |
активные гены, кодирующие |
белки, |
обслужи |
|||
|
|
вающие базовые функции всех клеток организма, противопос |
|||||
|
|
тавляются "luxury" генам—генам |
"роскоши", |
обслуживающим |
|||
|
|
специфические функции дифференцированных клеток |
|||||
Гибридома |
— соматический |
гибрид антителообразующей |
клетки |
и клетки |
|||
|
|
миеломы (опухоли В-клеток), продуцирующий моноклональ |
|||||
|
|
ные антитела |
(МкАТ) |
|
|
|
|
ГК |
— гиалуроновая |
кислота |
|
|
|
||
ГМ - КСФ |
— грануло-моноцитарный колониестимулирующий фактор |
||||||
ГС |
— |
гепарансульфат |
|
|
|
|
|
ГСТ-П |
— плацентарная форма у-глютатион-5-трансферазы |
|
|||||
ГТФ . |
— |
гуанозинтрифосфат |
|
|
|
|
|
ГФРТ |
— |
гипоксантинфосфорибозилтрансфераза |
|
|
|||
ддм |
— |
4,4'-диаминодифенилметан |
|
|
|
||
ДДТ |
— |
дихлордифенилтрихлорэтан |
|
|
|
||
Д К |
— дендритные клетки |
|
|
|
|
||
15
Д М БА |
7,12-диметилбенз(а)антрацен |
||
Д М Г |
1,2-диметилгидразин |
|
|
Д Э С |
диэтилстильбэстрол |
|
|
ИД |
|
иммунодиагностика |
|
Идиотипические |
уникальные клоноспецифические детерминанты, локализован |
||
детерминанты Ig |
|||
|
|
ные в районе активного центра Ig\ |
|
ИЛ |
|
интерлейкины |
/ |
ИМ |
|
индометацин |
|
Иммунотоксин |
конъюгат моноклонального антитела с белковым токсином, |
||
предназначенный для поражения клеток-мишеней |
|||
Импринтинг |
гаметоспецифичная модификация Д Н К , в результате которой в |
||
соматической клетке осуществляется транскрипция только ма |
|||
|
|
теринского или только отцовского аллеля |
|
И П Ф Р |
инсулиноподобный фактор роста |
||
И Ф Н |
интерферон |
|
|
КА |
|
капсидный антиген EBV |
|
кб, |
kb |
тысяча пар оснований, |
kilobases |
кДа |
(kD) |
килодальтон, единица измерения мол. массы белка |
|
Клетки HRS |
клетки Ходжкина—Рида—Штернберга |
||
КОЕ |
колониеобразующая единица |
||
К О М Т |
катехол-О-метилтрансфераза |
||
КСТ |
краткосрочные тесты |
|
|
К С Ф |
колониестимулирующий |
фактор |
|
КТП |
канал трансмембранной |
проводимости |
|
ЛБ |
(BL) |
лимфома Беркитта |
|
ЛГ |
|
лютеинизирующий гормон (от латинского luteus—желтый) |
|
ЛКЛ |
лимфобластоидные клеточные линии |
||
лип |
|
липопротеин низкой плотности |
|
лпэ |
|
линейная передача энергии |
|
лт |
|
лимфотоксин |
|
ЛУ |
|
лекарственная устойчивость опухолевых клеток |
|
МАП К |
митогенактивированные |
протеинкиназы |
|
МДС |
миелодиспластический |
синдром |
|
Миссенс (missense) |
мутации гена, нарушающие смысловую последовательность |
||
МкАТ |
моноклональные антитела—антитела, продуцируемые одним |
||
|
|
клоном В-лимфоцитов, характеризующиеся абсолютной одно |
|
|
|
родностью по всем иммунологическим и физико-химическим |
|
|
|
признакам |
|
М К П К |
мононуклеарные клетки периферической крови |
||
МЛУ |
множественная лекарственная устойчивость опухолевых клеток |
||
ММ |
|
множественная миелома |
|
ммс |
метилметансульфонат |
|
|
М Н И Ф |
метод непрямой иммунофлюоресценции |
||
М Н Н Г |
N-метил-N-нитронитрозогуанидин |
||
Мониторинг |
прослеживание динамики развития опухолевого клона |
||
М ф |
|
макрофаги |
|
М Щ К |
межклеточные щелевые |
контакты |
|
н |
|
нуклеотиды |
|
НБГПА |
N-нитрозобис(2-гидроксипропил)амин |
||
НДЭА |
N-нитрозодиэтиламин |
|
|
Н М М |
N-метил-N-нитрозомочевина |
||
Н М М |
нитрозометилмочевина |
|
|
H C C |
гепатоцеллюлярная карцинома |
||
Н ф |
|
нейтрофилы |
|
нхл |
неходжкинская лимфома |
||
ОБЭ |
относительная биологическая эффективность |
||
ОКГ |
основной комплекс гистосовместимости |
||
ОЛ |
|
острый лейкоз |
|
16
ОЛЛ |
— острый лимфобластный лейкоз |
|
|
О М Ж |
— |
опухоли молочных желез |
|
ОМЛ |
— острый миелобластный лейкоз |
|
|
ОНЛЛ |
— острый нелимфобластный лейкоз |
|
|
ОПЛ |
— острый промиелоцитарный лейкоз |
|
|
ОР |
— относительный риск |
|
|
п.н. |
— |
пары нуклеотидов |
|
ПАР |
— превышение абсолютного риска, excess absolute |
risk |
|
Паттерн |
|
\ |
|
метилирования |
— расположение в Д Н К неметилированных и метилированных |
||
|
|
CpG-сайтов, характерное для данного типа клеток |
|
ПАУ |
— |
полициклические ароматические углеводороды |
|
ПГ |
— |
протеогликаны |
|
ПГЕ |
— простагландины Е типа |
|
|
ПД50 |
— 50 % прививочная доза опухолевых клеток |
|
|
ПДД |
— предельно допустимые дозы канцерогенов |
|
|
ПДК |
— предельно допустимые концентрации канцерогенов |
||
Плазмоцитомы |
— |
опухоли плазматических клеток |
|
ПР (PR) |
— |
протеаза |
|
Провирусная нагрузка — число мононуклеарных клеток периферической |
крови, содер |
||
|
|
жащих провирус (например, HTLV-1) |
|
ПСА |
— специфический антиген простаты |
|
|
ПСМА |
— специфический мембранный антиген простаты |
|
|
П Ц Р (PCR) |
— полимеразная цепная реакция |
|
|
РА |
— ранний антиген EBV |
|
|
РЖ |
— рак желудка |
|
|
РКС |
— фосфокиназа С |
|
|
РМЖ |
— рак молочной железы |
|
|
РНГ (NPC) |
— рак носоглотки |
|
|
РЭА |
— раково-эмбриональный антиген, серологический маркер опу |
||
|
|
холей прямой и толстой кишки |
|
Сингенные животные |
— животные (обычно мыши инбредных линий), |
идентичные по |
|
|
|
антигенам тканевой совместимости опухолям, возникшим в |
|
|
|
этой линии |
|
СК |
— саркома Капоши (Kaposi's sarcoma, KS) |
|
|
СК II |
— казеинкиназа II |
|
|
СОД |
— |
супероксиддисмутаза |
|
СПИД |
— синдром приобретенного иммунодефицита человека |
||
СПП |
— стимуляторы пролиферации пероксисом |
|
|
СТОА |
— специфические трансплантационные опухолевые антигены |
||
СХО |
— сестринские хроматидные обмены |
|
|
СЭМ |
— сканирующая электронная микроскопия |
|
|
т. п.н. |
— |
тысяча пар нуклеотидов |
|
ТБ |
— |
терминальный-белок |
|
ТК |
— тимидинкиназа |
|
|
ТМ |
— трансмембранный белок |
|
|
ТФА |
— |
тетрадеканоилфорболацетат |
|
ТФР |
— трансформирующий фактор роста бета |
|
|
Тх-1, 2 |
— Т-хелперы двух классов |
|
|
УФ-облучение |
— |
ультрафиолетовое облучение |
|
ФАНФТ |
— Л'-[4-(5-нитро-2-фурил)-2-тиазолил]формамид |
|
|
ФБ |
— |
фенобарбитал |
|
ФИЛ |
— фактор ингибиции лейкоцитов |
|
|
ФН О — фактор некроза опухолей
ФП Э — феномен "переключения эстрогенного эффекта" ФРФ-2 — основной фактор роста фибробластов
ФСГ |
— |
фолликулостимулирующий гормон |
Х-4-С, Х-6-С |
— |
хондроитинсульфаты |
ХПЛ |
— хронический лимфолейкоз |
|
2 - 7908 Д. Г. Заридзе |
17 |
|
хмл хммл
Цитокины
ЦТА
ЦТД
ЦТЛ
Чекпойнт, checkpoint, "сверочная точка"
ЭБВ (EBV)
Экстравазация Э М С
Эпигенетическая
изменчивость
Эпитоп
ЭР
ЭР
ЭФ
ЭФ Р А20
АВС-семейство
AIDS
AIF
A M F
AML
anchoring junctions АР-1
АРС ARF
ATF
ATL
ATLL ATM ATR В7
BaEV Вах, Вас BCBL bcl-2
bcr
хронический миелоидный лейкоз хронический миеломоноцитарный лейкоз
низкомолекулярные пептиды, контролирующие активность клеток иммунной системы макрофагов, нейтрофиллов, В- и Т- лимфоцитов, NK-клеток и тучных клеток
цитотоксическая активность цитотоксическое действие цитотоксическике лимфоциты
любая стадия клеточного цикла, на которой цикл может быть остановлен и вход в следующую фазу отложен
вирус Эпштейна—Барр, HHV4 (human herpesvirus 4, герпесвирус человека 4-го типа)
выход клеток из сосудистого русла этилметансульфонат
наследуемое изменение признака, при котором генетический код остается неизменным. В основе эпигенетической изменчи вости лежат изменения конформации хроматина, обусловлен ные такими его модификациями, как метилирование ДНК, ацетилирование гистонов и др.
детерминантная группа антигена, распознаваемая индивиду альным антителом эндоплазматический ретикулум эстрогеновые рецепторы эмбриональные фибробласты эпидермальный фактор роста
клеточный ген; кодирует белок, обладающий способностью блокировать апоптоз семейство белков-транспортеров, имеющих АТФ-связывающие сайты (ATP-binding cassette)
acquired immune deficiency syndrome, синдром приобретенного иммунодефицита
apoptosis inducing factor
autocrine motility factor, аутокринный фактор подвижности гепатоцитов / рассеивающий фактор острый миелоидный лейкоз заякоривающие контакты
activated protein 1, транскрипционный фактор, являющийся димером, содержащим по одному онкобелку от представителей семейств Jun и Fos
Adenomatous polyposis coli
alternative reading frame, альтернативная рамка считывания, pl4ARF— продукт альтернативной рамки считывания активирующий транскрипционный фактор
Т-клеточный лейкоз взрослых, ассоциированный с HTLV-I adult T-cell leukemia/lymphoma, Т-клеточный лейкоз взрослых ataxia-teleangictasia mutated
ataxia-teleangictasia related
костимуляторная молекула, присутствие которой на мембране антигенпрезентирующей клетки необходимо для активации Т- клетки
babuin endogenous retrovirus, эндогенный ретровирус бабуинов протоапоптотические белки семейства bcl-2
body cavity based lymphoma, лимфома полостей тела протоонкоген, активируемый хромосомной транслокацией в В- клеточной лимфоме человека. Продукт гена ингибирует апоптоз breakpoint cluster region, район 22-й хромосомы, включающий ся в филадельфийскую хромосомную транслокацию
18
BCR/ABL |
— белок слитный |
BCR-ABL |
— химерный ген |
BL (ЛБ) |
— лимфома Беркитта |
BLV |
— bovine leukemia virus, вирус лейкоза крупного рогатого скота |
BRCA-1, -2 |
— breast cancer associated, дйа гена-супрессора. ассоциированные |
|
с карциномой молочной железы |
САК |
— cyclin-dependent kinase-activating kinase, киназа, которая акти |
|
вирует cdks, фосфорилируя их |
CALLA |
— common acute lymphocytic leukemia antigen, общий антиген ост |
|
рых лимфатических лейкозов |
сАМР |
— циклический аминозинмонофосфат |
CARD |
— caspase recruitment domain (домен для рекрутирования каспаз) |
CASE или MultiCASE |
— computer automated structure evaluation, типы обучаемых про |
|
грамм |
СВР |
— CCAAT-binding factor |
СВР |
— CREB-взаимодействующий белок |
CD31 |
— молекула адгезии |
CD-I |
— синдекан-1 |
CD3 |
— комплекс молекул, необходимых для передачи активирующего |
|
сигнала от TCR в клетку |
CD4 |
— маркер Т-хелперов |
CD4 и CD8 |
— cluster differentiation 4 and 8, корецепторы TCR, взаимодейст |
|
вующие соответственно с презентирующей молекулой М Н С - П |
|
или M H C - I |
CD44 |
— молекула адгезии |
CD45 |
— поверхностный фосфат |
CD8 |
— маркер Т-супрессоров |
cdc |
— гены цикла клеточного деления |
cdk |
— циклинзависимые киназы |
CDKI |
— cyclin dependent kinase inhibitors |
cdks, или cdc 2 |
— cyclin-dependent kinases, циклинзависимые киназы |
c-FLIP |
— cellular FLIC E inhibitory protein (клеточный ингибитор кас |
|
паз) |
CKls |
— cyclin-dependentkinase inhibitors |
CMC |
— chemical mismatch cleavage, расщепление неспаренных основа |
|
ний |
CML |
— хронический миелолейкоз |
CMV |
— cytomegalovirus, цитомегаловирус, HHV5 (human herpesvirus 5, |
|
герпесвирус человека 5-го типа) |
cORF |
— central open reading frame, центральная открытая рамка считы |
|
вания |
СОХ |
— циклооксигеназа |
СОХ-1, СОХ-2 |
— изоформы циклооксигеназы |
CRE |
— циклический АМР-респонсивный элемент |
CREB |
. — белок, взаимодействующий с CRE-областью |
Csk |
— специфическая киназа |
СТЕ |
— constitutive transport element, базовый транспортный элемент |
CTL |
— цитотоксические Т-лимфоциты |
DAG |
— 1,2-диацилглицерин |
DcR |
— decoy receptor (рецептор, не способный участвовать в передаче |
|
сигнала) |
DD |
— death domain (домен смерти) |
DED |
— death effector domain (эффекторный домен смерти) |
desmosomes |
— десмосомальные межклеточные контакты |
differential display |
— дифференциальный дисплей м Р Н К на основе П Ц Р |
DISC |
- D E A T H - I N D U C I N G SIGNALING COMPLEX (комплекс, ин |
|
дуцирующий сигнал смерти) |
DNA-PK |
— ДНК-зависимая протеинкиназа |
downstream |
— более удаленное расположение фрагментов Д Н К или Р Н К и |
2* |
19 |
|
|
|
более поздняя их транскрипция или трансляция по отношению |
||
|
|
|
к какому-либо сайту |
|
|
DR |
|
|
death receptor (рецептор смерти) |
||
E |
|
|
ранние (early) транскрипты |
||
El A, E1B, E2A, E2B, |
|
|
|
||
ЕЗ и E4 |
|
|
ранние (early) области |
генома |
|
EA |
|
|
early antigen, ранний антиген |
||
EBER-1, -2 |
|
Epstein—Barr virus encoded RNA, малые неполиаденилирован- |
|||
|
|
|
ные РНК, кодируемые EBNA—Epstein—Barr virus nuclear anti |
||
|
|
|
gen 1, 2, ЗА, ЗВ, 3C, LP (leader protein), ядерные белки EBV— |
||
|
|
|
Epstein—Barr virus, герпесвирус Эпштейна—Барр |
||
ECM |
|
|
extracellular matrix, внеклеточный матрикс |
||
E G F |
|
|
epidermal growth factor, эпидермальный фактор роста |
||
E G F |
|
|
эпидермальный фактор роста |
||
electronic substruction |
электронное |
вычитание |
|||
ERV |
|
|
endogenous retrovirus, эндогенный ретровирус |
||
EST |
|
|
expressed sequence tag, |
вид анализа Д Н К |
|
Fab |
|
|
antigen binding fragment, фрагмент антител, реагирующий с де- |
||
|
|
|
терминантной группой антигена |
||
FAB |
|
|
франко-американо-британская классификация гемобластозов |
||
FADD-FAS |
|
associated death domain protein (белок, содержащий домен смер |
|||
|
|
|
ти и ассоциированный с FAS-рецептором) |
||
РАК |
|
|
тирозиновая протеинкиназа фокальных контактов |
||
P G P |
|
|
fibroblast growth factor, фактор роста фибробластов |
||
FISH |
|
|
fluorescein in situ hybridisation, флюоресцентная in situ гибриди |
||
|
|
|
зация |
|
|
Fp, ori-P, Cp, Wp |
|
регуляторные домены |
(промоторы) EBV |
||
gain-of-function |
|
|
|
|
|
мутации |
|
|
мутации, которые повышают нормальный уровень активности |
||
|
|
|
продукта гена, например онкогенные мутации в генах, вклю |
||
|
|
|
ченных в контроль роста |
||
gap junctions |
|
щелевые межклеточные контакты |
|||
H и L |
|
|
тяжелая и легкая цепи иммуноглобулинов соответственно |
||
HBV |
|
|
hepatitis virus В, ДНК-содержащий вирус сывороточного гепа |
||
|
|
|
тита типа В |
|
|
Hcs |
|
|
hepatocarcinogen sensitivity, локус |
||
HCV |
|
|
вирус гепатита С |
|
|
HeLaV |
|
|
вирус из клеточной линии HeLa |
||
HEp-2V |
|
|
вирус из клеточной линии НЕр-2 |
||
HERV |
|
|
human endogenous retrovirus, эндогенный ретровирус человека |
||
HETE |
|
|
липоксигеназные метаболиты АК (5-, 12- и 15-гидроксиэйкоза- |
||
|
|
|
пентоеновые кислоты) |
||
HFV |
|
|
human Faomy virus, Вирус Фаоми человека |
||
HHV-1 |
|
|
|
|
|
(human |
herpesvirus |
1) |
герпесвирус |
человека |
1-го типа |
HHV-2 |
|
|
|
|
|
(human |
herpesvirus |
2) |
герпесвирус |
человека |
2-го типа |
HHV-4 |
|
|
|
|
|
(human |
herpesvirus |
4) |
герпесвирус |
человека |
4-го типа |
HHV-6, HHV-7, |
|
|
|
|
|
HHV-8 |
|
|
human herpesvirus type 6, 7, 8, герпесвирус человека 6-, 7-, 8-го |
||
|
|
|
типов; относится к подсемейству у-герпесвирусов, роду Ради- |
||
|
|
|
новирусов |
(Rhadinoviruses) |
|
HIV |
|
|
human immunodeficiency virus, вирус иммунодефицита человека |
||
HLA |
|
|
система лимфоцитарных антигенов человека |
||
H M L |
|
|
human MMTV-like |
|
|
HOC |
|
|
остеосаркома человека |
||
H P |
|
|
Helicobacter |
Pylori |
|
HPLC |
|
|
high performance liquid chromatography |
||
20
HPV
HSV-1
HSV-2
HTDV
HTLV-I, HTLV-II
HVS
HERV
IAP
IDDM
IFA
IFN
Ig
IL-2
IL-2R
IL-6
IN
INK
IRES
JAK
Jk (CBF1), PU.l, TFIIB, TAF40, TFIIH JNK
JSRV
KSHV
L
L и S
LI—L5
LAK-клетки
LANA (ORF 73)
LC-y
LCR
LINE
LMP
LNA (ORF 73)
LOH
LOX
LTR
LUR
LV
MA
Macl (CDI lb/CD 18) MAGE и BAGE
MALT
MAP
MAP kinase
—human papilloma viruses, вирусы папиллом человека
—herpes simplex virus 1, вирус простого герпеса 1-го типа
—herpes simplex virus 2, вирус простого герпеса 2-го типа
—human teratocarcinoma-derived virus
—human T-cell leukemia virus, types I, II, вирус Т-клеточного лей коза человека 1-го или 2-го типа
—herpesvirus saimiri, герпесвирус саймири
—human endogenous retrovirus, эндогенные ретровирусные после довательности человека
—intracisternal type A particles, интерстициальные частицы типа А
—insulin dependet diabet mellitus, инсулинзависимый диабет 1-го типа
—immunofluorescent assay, метод иммунофлюоресценции
—интерферон
—иммуноглобулин
—интерлейкин-2
—рецептор интерлейкина-2
—интерлейкин-6
—интеграза
—inhibitor of kinase
—internal ribosome entry site, дополнительные области посадки рибосом
—Janus kinase
—клеточные транскрипционные факторы
—семейство киназ, включенных во внутриклеточные сигнальные каскады
—Jaagsiekte, вирус овец
—Kaposi's sarcoma herpesvirus, герпесвирус, ассоциированный с
СК, HHV-8 |
' |
—поздние (late) транскрипты
—long и short, два аллеля гена L-myc
—поздние (late) области
—lymphokin-activated killers
—ядерный антиген, ассоциированный с латентным состоянием HHV-8
—фосфолипаза С-у
—long control region, регуляторная область
—long interspread nucleotide element, длинный диспергированный повтор
—Latent membrane protein 1, 2A, 2B, латентные мембранные бел ки EBV
—ядерный антиген латентного цикла HHV-8, являющийся ком понентом
— loss of heterozigosity, потеря гетерозиготное™
—липоксигеназа
—long terminal repeat, длинный концевой (терминальный) повтор
—long unique repeats, длинные уникальные последовательности
—вирус лангура
—белки матрикса
—антигены дифференцировки лейкоцитов человека
—семейства генов, экспрессирующихся в яичке и различных опу холях и контролирующих опухолевые антигены, активирующие цитотоксические Т-клетки
—mucosa associated lymphoid tissue, лимфоидная ткань слизистой оболочки
—mitogen activated protein kinases, митогенактивированные протеинкиназы
—mitogen-activated protein kinase (ERK—externally regulated kinase)
21
M C D |
— |
multicentric Castelman's disease, мультицентрическая болезнь |
||||
|
|
Кастельмана |
|
|
|
|
MDC |
— |
ген металлопротеазы |
|
|
|
|
M D S |
— |
миелодиспластический синдром |
|
|
||
MeCbl |
— метилкобаламин, кофермент метионинсинтазы |
|
||||
M H C I и М Н С II |
— major histocompatibility complex |
I or II, |
главный комплекс гис- |
|||
|
|
тосовместимости I и II классов, белковые молекулы, контроли |
||||
|
|
руемые генами тканевой совместимости и презентирующими |
||||
|
|
фрагменты антигена Т-клетками |
|
|
||
mlg |
— моноклональные Ig, продуцируемые опухолями антителообра- |
|||||
|
|
зующих |
клеток |
|
|
|
mismatch repair |
— репарация неспаренных оснований |
|
|
|||
MIL |
— myeloid lymphoid leukemia, ген |
|
|
|
||
MMTV |
— mouse mammary tumor virus, |
вирус |
рака молочной |
железы |
||
|
|
мыши |
|
|
|
|
MoMuLV |
— Molony mouse leukemia virus, вирус лейкоза мышей Молони |
|||||
M R P |
— multidrug |
resistance associated |
protein, |
белок, определяющий |
||
|
|
один из типов множественной лекарственной устойчивости |
||||
M S F |
— migration stimulating factor, фактор, стимулирующий миграцию |
|||||
N C |
— нуклеокапсидный белок |
|
|
|
||
NES |
— nucleus export signal, сигнал ядерного экспорта |
|
||||
NFkB |
— ядерный транскрипционный комплекс кВ . |
|
||||
NK-клетки |
— natural killers, естественные киллеры |
|
|
|||
NLS |
— nucleus localization signal, сигнал локализации (белка) в ядре |
|||||
N O E L |
— no observable effect level, максимальная неэффективная доза |
|||||
NAT-2 |
— К-ацетилтрансфераза-2 |
|
|
|
||
N C |
— нуклеокапсидный белок |
|
|
|
||
occluding, |
|
|
|
|
|
|
tight junctions |
— замыкающие, ил» плотные межклеточные контакты |
|
||||
O R F |
— open reading frame, открытая рамка считывания |
|
||||
O R F 25, O R F 26, |
|
|
|
|
|
|
O R F 65 |
— гены, кодирующие белки литического цикла |
|
||||
O R F 72 |
— вирусный ген, кодирующий |
белок-гомолог циклинов |
типа Д |
|||
|
|
млекопитающих |
|
|
|
|
O R F 74 |
— вирусный ген, кодирующий белок-гомолог рецептора хемокина |
|||||
|
|
человека |
|
|
|
|
O R F BALF5, O R F BALF2, |
|
|
|
|
||
O R F BORF2, O R F BARF1, |
|
|
|
|
||
O R F BXLFI и др. |
— гены, транскрибирующиеся на ранних этапах EBV-инфекции |
|||||
O R F BARFO |
— ген, кодирующий м Р Н К в клетках, латентно инфицированных |
|||||
|
|
EBV |
|
|
|
|
O R F BLLF1, O R F BALF4, |
|
|
|
|
||
O R F BXLF2, O R F BZLF2, |
|
|
|
|
||
O R F BCRF1, O R F BHRFI |
|
|
|
|
||
и др. |
— гены, кодирующие гликопротеины позднего цикла |
EBV-ин |
||||
|
|
фекции |
|
|
|
|
O R F cLFl, O R F N R F 1 , |
|
|
|
|
|
|
O R F BXRF1, |
|
|
|
|
|
|
O R F BFRF3 |
— гены, кодирующие негликозилированные белки позднего цик |
|||||
|
|
ла EBV-инфекции |
|
|
|
|
O R F Kl |
— вирусный ген, являющийся потенциальным онкогеном HHV-8 |
|||||
Ori-P |
— точка инициации репликации |
|
|
|
||
P450 (CYP) |
— |
семейство цитохромов |
|
|
|
|
p53 |
— супрессорный ген, кодирующий белок р53 |
|
||||
P53 |
— ядерный |
фосфопротеин, нормальная |
функция которого кон |
|||
|
|
тролировать клеточную пролиферацию. Мутация или отсутст |
||||
|
|
вие белка ассоциируется с некоторыми опухолями |
|
|||
PA |
— ревматоидный артрит |
|
|
|
||
Papovaviridae |
— papilloma-polyoma—vacuolating viruses |
|
|
|||
22
РВМС |
— мононуклеарные клетки периферической крови |
|
|
|
|
||||
PCNA |
— proliferating cell nuclear antigen, |
субъединица |
ДНК - полиме - |
||||||
|
|
разы |
|
|
|
|
|
|
|
PCR |
— polymerase chain reaction, полимеразная цепная реакция, П Ц Р |
||||||||
P D G F |
— platelets derived growth factor, тромбоцитарный фактор роста |
|
|||||||
P D G F / ш |
— фактор роста тромбоцитов—прототип вирусного онкогена sis |
||||||||
PEL |
— primary effusion lymphoma, первичная выпотная лимфома |
|
|
||||||
PG |
— |
простагландины |
|
|
|
|
|
|
|
Pgp |
— |
Р-гликопротеин |
|
|
|
|
|
|
|
Pgp-МЛУ |
— множественная лекарственная устойчивость, обусловленная |
Р- |
|||||||
|
|
гликопротеином |
|
|
|
|
|
|
|
Ph1 |
— филадельфийская |
хромосома, |
результат транслокации |
фраг |
|||||
|
|
мента 22-й хромосомы на хромосому 9 |
|
|
|
|
|||
PI3K |
— фосфатидилинозитол-3-киназа |
|
|
|
|
|
|
||
Р1Р2 |
— фосфатидилинозлтол-4,5-дифосфат |
|
|
|
|
||||
PMFV |
— вирус из клетбчной линии P M F |
|
|
|
|
|
|||
PMV |
— обезьяний вирус Мезон—Пфайзера |
|
|
|
|
||||
Ро-1-Lu |
— |
вирус очкового лангура |
|
|
|
|
|
|
|
PPAR |
— peroxisome proliferator activated receptor |
|
|
|
|
||||
PRb |
— белок, кодируемый |
геном Rb |
и |
являющийся |
опухолевым |
су- |
|||
|
|
прессором |
|
|
|
|
|
|
|
PRINS |
— |
П Ц Р in situ на хромосомных препаратах |
|
|
|
|
|||
PSA |
— prostate specific antigen, серологический антиген рака простаты |
||||||||
РТНгР |
— паратироидноподобный белок |
|
|
|
|
|
|
||
PTLV |
— группа Т-лимфотропных вирусов приматов |
|
|
|
|
||||
рТуг |
— фосфорилированный тирозин |
|
|
|
|
|
|
||
РТВ |
— фосфотирозинсвязывающий домен |
|
|
|
|
||||
QSAR |
— quantitative structure activity relationship, соотношение |
структу |
|||||||
|
|
ра/функция для прогноза канцерогенности |
|
|
|
|
|||
RA |
— ретиноевая кислота |
|
|
|
|
|
|
||
RA |
— рефрактерная анемия |
|
|
|
|
|
|
||
RAEB |
— рефрактерная анемия с избытком бластов |
|
|
|
|
||||
RAEB-T |
— рефрактерная анемия с избытком |
бластов в стадии трансфор |
|||||||
|
|
мации |
|
|
|
|
|
|
|
RAR |
— ген рецептора ретиноевой кислоты |
|
|
|
|
||||
RAR, RXR |
— рецепторы ретиноевой кислоты |
|
|
|
|
|
|
||
RARA |
— ген, кодирующий рецептор ос-ретиноевой кислоты |
|
|
|
|||||
RARb |
— респонсивный ген ретиноевой кислоты |
|
|
|
|
||||
RAS |
— рефрактерная сидеробластная анемия |
|
|
|
|
||||
Rb |
— ген ретинобластомы (клеточный антионкоген) |
|
|
|
|
||||
RD |
— рабдомиосаркома |
|
|
|
|
|
|
|
|
RD-114 |
— ретровирус типа С кошек |
|
|
|
|
|
|
||
RER+ |
— replication error, наличие микросателлитных аллелей |
аномаль |
|||||||
|
|
ной длины |
|
|
|
|
|
|
|
Retroviridae |
— ретровирусы; название семейства |
происходит |
от англ. |
reverse |
|||||
|
|
transcriptase (обратная транскриптаза—фермент, |
входящий |
в |
|||||
|
|
состав ретровирусных вирионов) и лат. retro—назад, |
указывает |
||||||
|
|
на обратный поток информации от Р Н К к Д Н К |
|
|
|
||||
RFLP |
— restriction fragment lenght polymorfism, анализ полиморфных ал |
||||||||
|
|
лелей, ПДРФ-анализ |
|
|
|
|
|
|
|
RNP |
— |
ribonucleotide protein, рибонуклеопротеид |
|
|
|
|
|||
RRE |
— Rev-responsive element, Rev-зависимый элемент |
|
|
|
|
||||
RT |
— обратная транскриптаза |
|
|
|
|
|
|
||
RT-PCR |
— обратная П Ц Р ' |
|
|
|
|
|
|
|
|
SAG, Sag |
— |
superantigen, суперантиген |
|
|
|
|
|
|
|
SAIDS |
— синдром приобретенного иммунодефицита обезьян |
|
|
|
|||||
SF, или H G F / S F , |
— scatter factor, "рассеивающий фактор", или фактор роста |
|
|
||||||
SFV-1 |
— simean Faomy virus, вирус Фаоми обезьян |
|
|
|
|
||||
SH2-AOMCH |
— |
домен src-homology 2 |
|
|
|
|
|
|
|
23
SINE
SIV
SKY
SLE
SMRV
SRE
SRV
SRV-Pc
SS
SSAV
SSCP
STAT
STLV-I, STLV-II
STS
SU substructive hybridization
TBG
ТВР Tell TCR tet TGF - pi
TIL-клетки T I M P
TLR ТМ
T N F R 1
T N F - a
TRAIL
TSP/HAM Т-антигены URR
V, D и J
v-bcl-2 (ORF 16) VGA
v-cyclin D V E G F
v-IL 6 v-IRF
v-MIP-I, v-MIP-Il
VNTR
VZV
V-антигены
WAFl/p21
WT1
—short interspread nucleotide element, короткий диспергирован ный повтор
—вирус иммунодефицита обезьян
—многоцветное спектральное кариотипирование
—system lupus erythematosus, системная красная волчанка
—вирус беличьих обезьян
—сывороточный респонсивный элемент
—ретровирус типа D обезьян
—ретровирус типа D павианов
—Sjorgren's syndrom, синдром Сьегрена
—sarcoma simean associated virus, саркомаассоциированный вирус обезьян —
—single strand conformation polymorphism, анализ конформационного полиморфизма однонитевой Д Н К
—signal transducers and activators of transcription, активаторы транскрипции
—simian T-cell leukemia virus, type I, II, вирус Т-клеточного лей коза обезьян первого или второго типа
— sequence tagget sites, вид анализа Д Н К
—поверхностный белок вирусной оболочки
—разностная гибридизация SV40—simian virus 40, обезьяний ви рус 40
—thyroxin binding globulin,' белок, связывающий гормон щитовид ной железы
—TATA-binding protein
—транскобаламин II
—T-cell receptor, антигенраспознающий рецептор Т-клеток
—тетрациклинзависимый
—опухолевый ростовой фактор (51
—tumor-infiltrating lymphocytes
—тканевый ингибитор металлопротеиназ
—TOLL-like receptor
—трансмембранный белок вирусной оболочки
—Tumour Necrosis Factor Receptor 1 (рецептор фактора некроза опухоли 1)
—фактор некроза опухолей альфа
— TNF-related apoptosis-inducing ligand (лиганд из семейства
Ф Н О , индуцирующий апоптоз)
—тропический спастический парапарез
—опухолевые антигены (on tumor), вирусспецифические белки
—upstream regulatory region, регуляторная область
—фрагменты генов Н-цепи (V, D и J) и легкой цепи (V, J)
—вирусный ген, гомологичный клеточному гену bcl-2
—virus capsid antigen, вирусный капсидный антиген
—вирусный гомолог D2 циклина человека
—фактор роста эндотелия сосудов
—белок, вирусный гомолог интерферона-6 человека
—O R F К9, белок, вирусный гомолог интерферона человека
—белки (кодируемые O R F Кб и O R F K4J соответственно), яв ляющиеся гомологами белков макрофагального воспаления и MIP-II
—variable nucleotide tandem repeates
—Varicella-zoster virus, вирус зостер, HHV3 (human herpesvirus 3, герпесвирус человека 3-го типа)
—вирионные белки
—универсальный ингибитор циклинзависимых киназ
- Wilms' Tumor 1
Введение
Канцерогенез — сложный много стадийный процесс, для реализации которого необходимо несколько", по следовательных генетических собы тий. Злокачественная опухоль разви вается в результате клональной экс пансии клеток, которые приобретают селективное преимущество в росте вследствие одного или нескольких из менений в геноме клетки.
Тот факт, что злокачественные опухоли редко возникают de novo и что им предшествуют предраковые из менения, из которых с той или иной степенью вероятности может развить ся злокачественная опухоль, был из вестен давно. В результате гистологи ческих и цитологических исследова ний и наблюдений за больными с доброкачественными опухолями и пролиферативными, диспластическими поражениями, как, например, аденоматозные полипы толстой кишки и дисплазия шейки матки, было показа но, что из них с высокой степенью ве роятности развивается интраэпителиальный, а затем и инвазивный рак.
Экспериментальные исследова ния, касающиеся индукции опухолей с применением химических канцеро генов, привели к созданию в 60-х го дах концепции инициации и промо ции. Согласно этой концепции, в ре зультате инициации клетка претерпе вает необратимые изменения, кото рых, однако, недостаточно для ее пре вращения в опухолевую. На стадии же промоции в клетке происходят про цессы, приводящие к экспрессии опу холевого фенотипа, т. е. превращению инициированной клетки в опухоле вую. Процесс возникновения и отбора новых клонов опухолевых клеток в организме лежит в основе прогрессии опухоли (теория Foulds), идущей в
сторону усиления злокачественных свойств опухолевых клеток. Прогрес сия включает автономизацию роста и возникновение метастатической ак тивности.
Открытие генов и их продуктов, участвующих в регуляции пролифера ции и дифференцировки клеток и иг рающих ведущую роль в формирова нии опухоли, стало переломным эта пом в понимании механизмов канце рогенеза. К этим генам в первую оче редь относятся протоонкогены и ге- ны-супрессоры, структурные измене ния, гиперэкспрессия или потеря ко торых приводит к нарушению контро ля нормального клеточного роста, дифференцировки и пролиферации и в конечном счете к злокачественной трансформации клетки. За последнее время были идентифицированы и клонированы десятки онкогенов и ге нов-супрессоров, а также других не входящих в эти две группы генов, ко торые участвуют в прогрессии и метастазировании опухоли. Злокачествен ная опухоль развивается в результате инакцивации генов-супрессоров и ак тивации онкогенов. Накопление этих изменений приводит к прогрессивно му изменению фенотипа клеток. На основании математического модели рования процесса канцерогенеза у че ловека было высказано предположе ние, что для превращения нормаль ной клетки в опухолевую требуется от 3 до 7 независимых случайных собы тий. Например, при раке толстой кишки происходит инактивация ге нов-супрессоров (АРС, МСС, р53 и DCQ и активация протоонкогена К- ras, причем некоторые из этих изме нений обнаруживаются на ранних ста диях, т. е. в аденоматозных полипах, а другие — на более поздних стадиях
25
канцерогенеза и прогрессии опухоли. Изучение наследственных синдро мов, таких как, например, семейная ретинобластома и аденоматозный полипоз толстой кишки, показало, что первой ступенью процесса канцероге неза могут быть наследуемые терми нальные мутации в генах Rb и АРС. Однако для развития злокачественных опухолей необходимы дополнитель ные генетические изменения в сома тических клетках. Вероятность воз никновения в одной клетке несколь ких генетических изменений повыша ется при нарушениях, ведущих к гене тической нестабильности, т. е. при нарушениях работы систем, поддер живающих целостность генома.
В развитии злокачественной опу холи важную роль играют и эпигене тические процессы, которые приводят к изменению фенотипа, но не сопря жены с нарушением последовательно стей ДНК. К ним относится метили рование ДНК. Ряд веществ, вызываю щих метилирование ДНК, являются доказанными канцерогенами для ла бораторных животных.
Пролиферация является необходи мым компонентом процесса канцеро генеза. Она может быть результатом генетических изменений в клетке, но может быть связана с другими физио логическими или патологическими процессами и предшествовать измене нию в геноме клетки. Репликация ДНК в пролиферирующих клетках де лает их более чувствительными к воз действиям, вызывающим поврежде ния их генома. В них также увеличи вается вероятность спонтанных мута ций, так что пролиферация эмпириче ски может быть охарактеризована как ранняя стадия канцерогенеза.
Важное значение в процессе кан церогенеза имеет приобретаемое трансформированными клетками свойство ухода от апоптоза, т. е. при сущего нормальной клетке механизма клеточного самоубийства. Уход от апоптоза повышает жизнеспособность трансформированной клетки, делает ее менее чувствительной к факторам
противоопухолевого иммунитета и те рапевтическим воздействиям. К важ нейшим приобретенным свойствам опухолевой клетки принадлежит их способность стимулировать неоангиогенез, т. е. формировать новые крове носные и лимфатические сосуды.
Открытие теломеразы — фермента, удлиняющего концы линейных хро мосом, и роли этого явления в про цессе формирования опухоли являет ся одним из наиболее значимых в об ласти фундаментальной онкологии. В связи с тем что примерно 85 % опухо лей человека обладают теломеразной активностью, можно утверждать, что реакция теломеразы участвует в кан церогенезе, следовательно, ингибирование (репрессия) теломеразы должно уменьшать вероятность развития опу холи.
Канцерогенные вещества принято делить на генотоксичные и негенотоксичные. Генотоксичные канцерогены взаимодействуют с клеткой чаще все го через метаболиты, которые ковалентно связываются с клеточными белками и ДНК и образуют аддукты. Описаны аддукты ДНК со многими канцерогенными веществами, а имен но с ПАУ, ароматическими аминами, N-нитрозосоединениями и афлатоксином В1. Аддукты канцерогена с ДНК отличаются друг от друга своими мутагенными и канцерогенными свойствами. Кроме того, важное^значение имеет концентрация того или иного аддукта в тканях-мишенях. Канцероген-ДНК-аддукты выводятся из организма спонтанно или в резуль тате репарации ДНК.
К негенотоксичным канцерогенам относятся вещества, неспособные ковалентно связываться с ДНК и обра зовывать аддукты. К ним относятся промоторы двухстадииного канцеро генеза, пестициды, гормоны, волок нистые материалы и т. д. Эти вещест ва, как правило, неспособны сами трансформировать клетки, и их канцерогенность скорее всего связана с созданием условий для преимущест венного роста клеток ранее трансфор-
26
мированных или спонтанно, или в ре зультате воздействия генотоксическо^ го канцерогена.
Причиной развития некоторых форм опухолей как у животных, так и у человека является вирусная инфек ция. Описано несколько возможных механизмов канцерогенеза, вызывае мого вирусами: а) протоонкоген со держится в вирусном геноме и интег рируется в клетку; б) вирус не содер жит онкогена, однако интеграция его геномного материала в клетку приво дит к нарушению регуляции экспрес сии клеточных генов; в) вирусный ге ном персистирует в виде эписомальных или интегрированных копий, и экспрессия вирусного гена приводит к активации клеточных последователь ностей, которые ответственны за про лиферацию. В результате эксперимен тальных и эпидемиологических иссле дований ряд вирусов признаны канце рогенными для человека.
Открытие феномена индивидуаль ной чувствительности к канцероген ным воздействиям, обусловленной на следственными особенностями орга низма, способствовало пониманию процесса канцерогенеза как результа та взаимодействия эндогенных и экзо генных факторов (Nature and Nurture). Генетические изменения, которые на следуются и с высокой вероятностью приводят к развитию рака, обычно выражаются в мутациях одного аллеля гена-супрессора. Идентифицирован ряд генов-супрессоров, врожденные мутации которых приводят к разви тию наследственных и семейных форм злокачественных опухолей. К ним относятся ген ретинобластомы (Rb), при мутации которого развива ется врожденная форма ретинобласто мы; ген-супрессор р53, врожденные мутации которого являются причиной синдрома первично-множественных опухолей Ли—Фраумени; ген аденоматозного полипоза толстой кишки (APQ и некоторые другие. Частота наличия этих мутаций среди населе ния крайне невелика, не более 1—5 случаев на 100 ООО живорожденных
младенцев. В то же время по признаку предрасположенности к развитию ра ка, которая определяется генами, от ветственными за метаболизм канцеро генных веществ, их активацию, деток сификацию и репарацию ДНК, насе ление полиморфно. Неблагоприятный фенотип может встречаться у 30— 50 % населения. Важную роль в мета болизме канцерогенных веществ игра ют ферменты цитохрома Р-450, кото рые активируют целый ряд канцеро генных веществ. Влияние на процесс канцерогенеза оказывают ферменты, которые определяют способность ДНК к репарации и репликации.
Теория, касающаяся роли иммуно логического статуса в развитии опухо лей человека, подтвердилась только для опухолей человека вирусного генеза — большинства злокачественных лимфом и саркомы Капоши. У боль ных, которым была произведена пере садка органа и которые получали иммуносупрессивную терапию, повышен риск развития злокачественной лимфомы и саркомы Капоши. Риск раз вития этих форм опухолей также чрез вычайно высок у больных с различны ми другими формами приобретенного иммунодефицита и в первую очередь у больных СПИДом и ВИЧ-инфици рованных.
Различие антигенной структуры опухолевых клеток и аутологичных нормальных клеток является основой для их распознания и элиминации двумя эффекторными системами за щиты организма против опухолей: а) врожденного иммунитета (естествен ного, неспецифического иммунитета) и б) приобретенного специфического противоопухолевого иммунитета. Эффекторные системы неспецифической естественной резистентности распо знают и отторгают опухолевые клетки независимо от экспрессии на них спе цифических опухолевых антигенов, в то время как распознание опухолевой клетки Т-лимфоцитами как "чужого" при развитии специфического проти воопухолевого иммунитета связано более всего со специфическими
27
трансплантационными опухолевыми антигенами или их аналогами, экспрессируемыми на клеточной мембра не. Специфически противоопухоле вый иммунитет связан с Т-клетками, распознающими опухолевые антиге ны, в тех случаях, когда эти антигены презентируются. В опытах in vitro бы ло установлено, что различные типы антигенов в опухолях человека дейст вительно могут распознаваться Т-лим- фоцитами, однако обнаружение неко торых из этих антигенов в нормаль ных органах и тканях человека, т. е. их тканеспецифический характер, от сутствие доказательств их участия в индукции противоопухолевого имму нитета делают сомнительными пер спективы успешного применения их в профилактике или лечении онкологи ческих больных.
Иммунорегуляторные сигналы воз никают при участии цитокинов, секретируемых эффекторными и опухо левыми клетками и работающих по типу локальных ростовых факторов позитивного и негативного действия. Получение рекомбинантных цитоки нов не только привело к настоящему прорыву в исследовании биологиче ской функции и передачи сигналов, но также сыграло важнейшую роль в клинической онкологии. Примене ние цитокинов можно рассматривать как один из вариантов неспецифиче ской иммунотерапии. Такие цитоки ны, как ИЛ-2, позволили культивиро вать линии и клоны цитотоксических
Т-клеток, узнающие опухолевые анти гены.
Исследования в области эпидемио логии рака показали, что факторы об раза жизни и окружающей среды яв ляются основной причиной развития 90—95 % злокачественных опухолей человека. По данным Международно го агентства по изучению рака, около 80 веществ, сложных смесей и про фессиональных факторов являются канцерогенными для человека. При этом причиной 30—35 % всех опухо лей человека является курение. Под тверждена роль онкогенных вирусов и некоторых бактерий и паразитов в этиологии рака. К доказанным канце рогенным факторам окружающей сре ды относятся ультрафиолетовая и ио низирующая радиация. В этиологии ряда злокачественных опухолей чело века важную роль играют факторы питания.
Таким образом, процесс канцеро генеза является результатом воздейст вия на человека множества различных факторов, как экзогенных, так и эн догенных. Переход от одной стадии в другую (последующую или предыду щую) также происходит в результате взаимодействия этих факторов, кото рые могут как способствовать, так и противодействовать этому процессу.
Предлагаемая вниманию читателей книга посвящена различным аспектам канцерогенеза и состоит из глав, осве щающих основные направления ис следований в этой области.
Г л а в а 1
ЭПИДЕМИОЛОГИЯ И ЭТИОЛОГИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ
Д.Г. Заридзе
1.1.Методологические зики, где деление каждого отдельного
подходы к изучению этиологии опухолей человека
Изучение этиологии болезней за нимает важное место как в медицин ской науке вообще, так и в онкологии в частности. Выявление этиологиче ских факторов опухолей человека яв ляется необходимой предпосылкой для их адекватной и эффективной профилактики, которая должна стро иться на научно обоснованной систе ме аргументов об этиологии опухолей человека.
Многовековой опыт изучения этиологии болезней показал, что при чина далеко не всегда реализуется в следствие, т. е. причинный фактор не всегда приводит к болезни, и что все поныне известные этиологические или причинные факторы имеют ха рактер "вероятностной", а не так на зываемой "строгой" причинности. Для реализации причины в следствие не обходим ряд дополнительных факто ров и условий. Казуальные отношения всегда многозначны, со многими из вестными и неизвестными.
Концепция "вероятностной" при чинности получила развитие в связи с разработкой теории вероятности и квантовой механики. Квантовомеханические процессы обладают принци пиально-статистическими свойства ми, что в пределах наблюдаемого на ми мира может вносить момент неоп ределенности в любые процессы. Не однозначность понятия причинности, необязательность реализации причи ны в следствие могут быть продемон стрированы примером из ядерной фи
атома может быть объяснено только с помощью теории вероятности.
Сказанное выше доказывает, что "строгая" причинность, т. е. обяза тельная реализация причинных фак торов в эффект, в результат, не един ственная форма причинно-следствен ной связи. Она соседствует с "вероят ностной" причинностью и последняя, по-видимому, является доминирую щей формой причинно-следственной связи как во всем объективном мире, так и в патологии.
Вероятностные процессы имеют статистический характер. Результаты таких процессов, как правило, невоз можно предсказать однозначно. Каж дое отдельное взаимодействие в стати стически детерминированном процес се относительно случайно.
Характерной особенностью подоб ного процесса является то, что он имеет место в дискретных системах, состоящих из большого числа элемен тов, относительно независимых друг от друга.
Концепция вероятностно-стати стической причинности чрезвычайно важна для правильного понимания, трактовки и оценки научных фактов, касающихся причин болезней. Отсут ствие "строгой" причинно-следствен ной связи между фактором и болез нью, когда причина не всегда реализу ется в болезнь, часто приводит к оши бочному отрицанию таковой как при чины.
Не вызывает сомнения, что мик роб, "возбудитель", является этиологи ческим фактором для того или иного инфекционного заболевания, однако так же несомненно, что заражение не
29
всегда приводит к болезни: для того |
ледованы (так называемые терминаль |
чтобы заражение микробом вызвало |
ные мутации), в этом случае роль |
болезнь, необходимо разрешение дру |
инициатора процесса • канцерогенеза |
гих дополнительных процессов. |
может играть унаследованная пред |
Открытие Ф. Леффлером бацилло |
расположенность. |
|
|
|
|
|
||||||||||||||
носительства и тот факт, что во время |
Кроме |
того, |
ряд |
унаследованных |
||||||||||||||||
даже сильных эпидемий (чума, холера |
характеристик внутренней среды орга |
|||||||||||||||||||
и оспа) не все инфицированные виру |
низма, как-то: способность метаболи- |
|||||||||||||||||||
лентными |
бактериями |
заболевали, |
зировать канцерогены и проканцеро- |
|||||||||||||||||
привело к признанию в инфекцион |
гены, репарировать ДНК — имеет |
|||||||||||||||||||
ной патологии роли внутренней реак |
важное значение в том, кто из экспо |
|||||||||||||||||||
ции организма. |
|
|
|
|
|
|
нированных заболеет раком. От цвета |
|||||||||||||
Аналогичная |
ситуация |
характерна |
кожи |
человека, |
например, |
зависит |
||||||||||||||
и для онкологии. Введение химиче |
чувствительность |
к |
канцерогенному |
|||||||||||||||||
ских канцерогенных |
веществ |
приво |
действию |
ультрафиолетовых лучей. |
||||||||||||||||
дит к возникновению опухоли не у |
|
На основании данных, полученных |
||||||||||||||||||
всех лабораторных животных в экспе |
за |
последнее |
десятилетие, |
можно |
||||||||||||||||
риментальной группе. |
|
|
|
|
предположить, |
что процесс |
канцеро |
|||||||||||||
Курение, |
которое, |
как |
известно, |
генеза — это постепенное накопление |
||||||||||||||||
является канцерогенным |
фактором |
мутаций и других изменений, приво |
||||||||||||||||||
для человека, вызывает рак далеко не |
дящих к активации протоонкогенов и |
|||||||||||||||||||
у всех курильщиков, хотя около 90 % |
инактивации или делении генов-су |
|||||||||||||||||||
всех случаев рака легкого вызваны ку |
прессоров в соматических клетках. На |
|||||||||||||||||||
рением. |
|
|
|
|
|
|
|
|
модели рака толстой кишки показано, |
|||||||||||
Доказано, что ряд химических ве |
что |
для |
превращения |
нормальной |
||||||||||||||||
ществ и производственных процессов, |
клетки в клетку злокачественной опу |
|||||||||||||||||||
ионизирующее |
и |
ультрафиолетовое |
холи необходимо, как минимум, пять |
|||||||||||||||||
облучение, курение, а также некото |
последовательных |
событий. |
|
|||||||||||||||||
рые вирусы и бактерии являются |
|
Степень |
выраженности |
вероятно |
||||||||||||||||
этиологическими факторами опухолей |
стно-статистической причинности в |
|||||||||||||||||||
человека, которые, однако, имеют ве |
медицине, в частности в онкологии, |
|||||||||||||||||||
роятностно-статистический |
характер. |
различна. |
Имеются |
примеры, когда |
||||||||||||||||
Это означает, что действие этих фак |
вероятностно-статистическая |
причин |
||||||||||||||||||
торов на человека не равнозначно за |
ность приближается к "строгой" при |
|||||||||||||||||||
болеванию |
злокачественными |
опухо |
чинности, следствие которой высоко |
|||||||||||||||||
лями. |
|
|
|
|
|
|
|
|
предсказуемо. |
Примером |
|
является |
||||||||
Для реализации эффекта канцеро |
значительная |
вероятность |
развития |
|||||||||||||||||
генного фактора, будь то химическое |
злокачественных опухолей у людей с |
|||||||||||||||||||
вещество или вирус, необходим ряд |
генетическими |
синдромами, |
такими |
|||||||||||||||||
дополнительных |
влияний, |
и |
конеч |
как синдром Ли—Фраумени, атаксия- |
||||||||||||||||
ный результат |
взаимодействия |
канце |
тел еангиэктазия, пигментная ксеро- |
|||||||||||||||||
роген—организм |
зависит от |
взаимо |
дерма, семейный полипоз и синдром |
|||||||||||||||||
действия ряда известных и неизвест |
Гарднера, |
нейрофиброматоз. Высокая |
||||||||||||||||||
ных экзогенных и эндогенных факто |
вероятность |
развития |
злокачествен |
|||||||||||||||||
ров. |
|
|
|
|
|
|
|
|
ных опухолей у людей с вышеуказан |
|||||||||||
Известно, что в результате канце |
ными генетическими синдромами мо |
|||||||||||||||||||
жет быть объяснена тем, что эти син |
||||||||||||||||||||
рогенных влияний |
окружающей сре |
|||||||||||||||||||
дромы, как и связанные с ними зло |
||||||||||||||||||||
ды возникают мутации |
и другие по |
|||||||||||||||||||
качественные |
опухоли, |
обусловлены |
||||||||||||||||||
вреждения в протоонкогенах и генах- |
||||||||||||||||||||
наследованием |
определенной генети |
|||||||||||||||||||
супрессорах, |
а |
также |
эпигенетиче |
|||||||||||||||||
ческой |
информации, |
а |
наследование |
|||||||||||||||||
ские изменения в соматических клет |
||||||||||||||||||||
генетической |
информации |
ввиду ее |
||||||||||||||||||
ках. |
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
высокой |
стабильности |
и |
предсказуе- |
|||||||||
Мутации |
в |
генах могут быть унас |
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||
30
мости приближается к "строгой" де терминации.
На теории вероятностно-статисти ческой причинности основана кон цепция факторов риска, получившая распространение в современной эпи демиологии.
Для практических целей эпидемио логам было предложено ввести в на учный лексикон понятия "достаточ ной", или "полной", причины, кото рой "достаточно" для возникновения заболевания, и причины, являющейся частью "полной" причины, "частич ной", которая лишь увеличивает веро ятность развития заболевания. При чинные факторы, изучаемые в эпиде миологии, являются "частичной" при чиной. Несмотря на то что влияние последней недостаточно для возник новения болезни, ее изъятие или бло кирование может привести к тому, что "полная" причина станет "непол ной", недостаточной для возникнове ния болезни, по крайней мере в пре делах одного определенного патогене тического механизма. В связи с этим для практической эпидемиологии, прежде всего в целях профилактики, знания о вероятностно-статистиче ских этиологических факторах опухо лей человека чрезвычайно важны, так как манипуляция этими факторами может привести к значительному сни жению заболеваемости злокачествен ными опухолями.
В эпидемиологии, как и во всех других научных дисциплинах, первым этапом исследования являются на блюдение и формулировка гипотезы. Гипотезы об этиологических факторах той или иной болезни эпидемиология черпает чаще всего из клинической практики и данных о распространен ности болезни у людей: в разных гео графических регионах, этнических, религиозных группах, концентрации того или иного заболевания в разных населенных пунктах, вокруг тех или иных предприятий, у работников раз ных профессий, лиц с различными бытовыми привычками, в определен ных семьях, у людей, болевших теми
или иными болезнями, и т. д. Источ ником возникновения гипотез о при чинах развития опухолей у человека также служат лабораторные исследо вания.
Эпидемиология неинфекционных болезней, частью которой является онкоэпидемиология, оперирует ря дом специфических методов, разрабо танных для изучения этиологических факторов болезней человека: описа тельные, аналитические и экспери ментальные.
Описательная, или дескриптивная, эпидемиология изучает распростране ние отдельных форм злокачественных опухолей в конкретных группах насе ления. Целью ее является описание особенностей распространения злока чественных опухолей. Предмет изуче ния описательной эпидемиологии — заболеваемость и смертность в разных географических регионах, населен ных пунктах, среди различных групп населения; связь (ассоциация) между различными характеристиками данно го региона, населенного пункта и на селения, например факторами окру жающей среды и образа жизни, и по казателями частоты злокачественных опухолей. Этот тип эпидемиологиче ского исследования принято называть
корреляционным, или экологическим. С
помощью этого метода можно опреде лить корреляцию между степенью вы раженности в данном регионе, среди данного населения того или иного фактора, в том числе факторов окру жающей среды и быта, и заболеваемо стью и смертностью от злокачествен ных опухолей. Однако возможности корреляционного метода в изучении причинно-следственных связей огра ничены. Корреляционный метод мо жет выявить связь между распростра ненностью того или иного фактора и частотой болезни среди данного насе ления, но не способен выявить связь между влиянием этого фактора и ин дивидуальным риском болезни.
Следующий шаг в постижении этиологического фактора — "сужение" ассоциации: переход от исследования
31
связи между изучаемым фактором и населением к связи между изучаемым фактором и конкретным человеком, который заболеет или заболел раком.
Такой подход к изучению этиоло гических факторов возможен с помо щью методов аналитической эпиде миологии, к ним относят:
1)проспективное, или когортное исследование;
2)ретроспективное исследование методом случай—контроль.
С помощью этих методов можно выявить связь между воздействием на человека предполагаемого этиологи ческого фактора и конкретным забо леванием.
Результаты этих исследований обычно выражаются в показателях от носительного риска (ОР), которые представляют собой соотношение за болеваемости группы экспонирован ного населения к заболеваемости группы неэкспонированного населе ния или соотношение заболеваемости населения с более выраженной экспо зицией к заболеваемости населения с менее выраженной экспозицией. По казатели ОР являются математиче ским выражением степени вероятно сти заболевания в результате воздей ствия определенного фактора.
Проспективное, или когортное, и ретроспективное исследования мето дом случай—контроль различаются тем, что в первом здоровое население, включенное в когорту для проспек тивного наблюдения, классифициру ется в зависимости от того, подверже но ли оно воздействию интересующего исследователей предполагаемого этио логического фактора, или от того, ка кова степень выраженности воздейст вия; при .ретроспективном исследова нии методом случай—контроль изучае мая группа населения классифициру ется на основании наличия или отсут ствия болезни. Обычно в исследование включаются больные и контрольная группа из той же группы населения.
Воздействие того или иного факто ра, степень его выраженности могут
быть охарактеризованы как на осно вании уже имеющихся гигиенических данных, так и с помощью их измере ния. Часто для оценки этого воздейст вия применяется анкетный метод.
Экспериментальные эпидемиологи ческие методы, или так называемые интервенционные контролируемые исследования, предполагают проведе ние эксперимента на человеке. В них обычно изучается эффект удаления или снижения воздействия предпола гаемого этиологического фактора на заболеваемость и смертность от опу холи, этиологически связанной с вы шеуказанным фактором. Имеется дру гой вид интервенционных исследова ний, когда включенные в исследова ние лица подвергаются влиянию фак торов с предположительно ингибирующим канцерогенез действием. За последние годы получили распростра нение интервенционные методы ис следования, с помощью которых изу чается роль витаминов, тамоксифена, аспирина и других возможных инги биторов канцерогенеза у человека.
Таким образом, эпидемиология, как всякая другая научная дисципли на, имеет в своем арсенале как эмпи рические, так и экспериментальные методы исследования, целью которых является выявление вероятностных причин или этиологических факторов опухолей человека. Фактор, будь то экзогенный или эндогенный, можно на зывать этиологическим только тогда, когда эпидемиологически доказана при чинно-следственная связь между воз действием фактора на человека и бо лезнью.
Необходимо отметить, что грубой методологической ошибкой является отнесение к связям этиологическим и ассоциативных. Выявление ассоциа тивных связей может привести к фор{ мулировке гипотезы о возможной этиологической роли того или иного фактора, но не служит ее доказатель ством. Как бы прочна ни была ассо циация, она всегда коренным образом отличается от причинных отношений.
Английский ученый Б. Хилл пред-
32
дожил ряд критериев, на основании ко |
ются достаточно обоснованные свиде |
||||||||||||||||||||||
торых можно дифференцировать при |
тельства их канцерогенности у лабо |
||||||||||||||||||||||
чинно-следственные связи от ассоциа |
раторных |
животных, |
с |
|
практической |
||||||||||||||||||
тивных. |
|
|
|
|
|
|
|
точки зрения должны рассматриваться |
|||||||||||||||
• |
Необходимым условием для оп |
как |
представляющие |
канцерогенную |
|||||||||||||||||||
опасность для человека. |
|
|
|
|
|||||||||||||||||||
|
ределения |
причинно-следствен |
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
ной |
связи |
является |
последова |
1.2. Дескриптивная |
|
|
|
|||||||||||||||
|
тельность в действии причины и |
|
|
|
|||||||||||||||||||
|
ее следствия: действия причины |
эпидемиология |
|
|
|
|
|
||||||||||||||||
|
или причинного фактора долж |
Заболеваемость |
и |
|
смертность |
от |
|||||||||||||||||
|
ны |
предшествовать |
следствию |
|
|||||||||||||||||||
|
злокачественных опухолей в мире изу |
||||||||||||||||||||||
|
или |
заболеванию. |
Кроме того, |
||||||||||||||||||||
|
чена достаточно хорошо. В большин |
||||||||||||||||||||||
|
необходимо принимать во вни |
||||||||||||||||||||||
|
стве |
развитых |
и ряде развивающихся |
||||||||||||||||||||
|
мание латентный период, необ |
||||||||||||||||||||||
|
стран |
принята |
система |
обязательной |
|||||||||||||||||||
|
ходимый для реализации канце |
||||||||||||||||||||||
|
регистрации |
всех |
вновь |
выявленных |
|||||||||||||||||||
|
рогенного воздействия того или |
||||||||||||||||||||||
|
случаев |
|
злокачественных |
опухолей. |
|||||||||||||||||||
|
иного фактора. |
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
Эта информация собирается в канцер- |
||||||||||||||||||
• |
Одним |
из |
главных |
показателей |
|||||||||||||||||||
регистре, |
|
где |
ее обрабатывают, под |
||||||||||||||||||||
|
возможной |
причинно-следст |
|
||||||||||||||||||||
|
считывают |
показатели |
заболеваемости |
||||||||||||||||||||
|
венной |
связи |
принято |
считать |
|||||||||||||||||||
|
всеми формами злокачественных опу |
||||||||||||||||||||||
|
выраженность связи между фак |
||||||||||||||||||||||
|
холей на территории данного канцер- |
||||||||||||||||||||||
|
тором |
и |
болезнью. |
Выражен |
|||||||||||||||||||
|
регистра и публикуют. |
|
|
|
|
|
|||||||||||||||||
|
ность этой связи |
определяется |
|
|
|
|
|
||||||||||||||||
|
Международное |
агентство |
по |
изу |
|||||||||||||||||||
|
величиной ОР. |
|
|
|
|
||||||||||||||||||
|
|
|
|
|
чению |
рака |
|
(МАИР) |
|
аккумулирует |
|||||||||||||
• |
Существенным |
критерием при |
|
|
|||||||||||||||||||
данные по заболеваемости |
из нацио |
||||||||||||||||||||||
|
чинно-следственной связи явля |
нальных |
|
регистров |
и |
публикует |
их |
||||||||||||||||
|
ется количественная связь меж |
|
|||||||||||||||||||||
|
1 раз |
в |
5 |
лет |
в виде статистического |
||||||||||||||||||
|
ду причиной и следствием по ти |
||||||||||||||||||||||
|
сборника "Рак на пяти |
континентах". |
|||||||||||||||||||||
|
пу "доза—эффект", т. е. от дозы |
||||||||||||||||||||||
|
Последнее (1997) издание этого спра |
||||||||||||||||||||||
|
(уровня |
и |
длительности влия |
||||||||||||||||||||
|
вочника |
|
содержит |
информацию, |
ос |
||||||||||||||||||
|
ния) зависит значение ОР. |
|
|||||||||||||||||||||
|
нованную на данных из 150 регистров, |
||||||||||||||||||||||
• |
Важным параметром для оценки |
||||||||||||||||||||||
расположенных в |
50 странах мира, и |
||||||||||||||||||||||
|
фактора с точки зрения причин |
||||||||||||||||||||||
|
дает |
характеристику |
|
заболеваемости |
|||||||||||||||||||
|
ной |
связи |
с болезнью |
является |
|
||||||||||||||||||
|
183 |
групп |
населения |
|
численностью |
||||||||||||||||||
|
подтверждение ассоциации в ря |
|
|||||||||||||||||||||
|
400 млн человек. |
|
|
|
|
|
|
|
|||||||||||||||
|
де исследований, в разных груп |
|
|
|
|
|
|
|
|||||||||||||||
|
Данные о смертности от злокачест |
||||||||||||||||||||||
|
пах населения, в различных ус |
||||||||||||||||||||||
|
венных опухолей, |
поступающие в от |
|||||||||||||||||||||
|
ловиях. |
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
дел статистики |
ВОЗ от подавляющего |
|||||||||||||||
• |
Очень важно, чтобы полученные |
||||||||||||||||||||||
большинства |
|
государств |
— |
членов |
|||||||||||||||||||
|
сведения |
о |
рассматриваемом |
|
|||||||||||||||||||
|
ВОЗ, охватывают большую часть насе |
||||||||||||||||||||||
|
этиологическом факторе не про |
||||||||||||||||||||||
|
ления мира. |
|
|
|
|
|
|
|
|
|
|||||||||||||
|
тиворечили тому, что известно о |
|
|
|
|
|
|
|
|
|
|||||||||||||
|
В |
табл. |
1.1 |
представлены |
сведения |
||||||||||||||||||
|
биологии и патологии |
данного |
|||||||||||||||||||||
|
о смертности от некоторых форм зло |
||||||||||||||||||||||
|
заболевания. Кроме того, жела |
||||||||||||||||||||||
|
качественных |
опухолей |
|
в |
45 |
странах. |
|||||||||||||||||
|
тельно, чтобы данные, получен |
|
|||||||||||||||||||||
|
В табл. |
|
1.2 |
показана |
заболеваемость |
||||||||||||||||||
|
ные в результате изучения болез |
|
|||||||||||||||||||||
|
основными формами злокачественных |
||||||||||||||||||||||
|
ни у человека, были подкрепле |
||||||||||||||||||||||
|
опухолей |
на |
основе |
канцер-регистров |
|||||||||||||||||||
|
ны |
результатами |
эксперимен |
||||||||||||||||||||
|
из 24 стран. |
|
|
|
|
|
|
|
|
|
|||||||||||||
|
тальных |
исследований. |
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
По данным МАИР, в 2000 г. злока |
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||||
При отсутствии |
эпидемиологиче |
чественными опухолями в мире забо |
|||||||||||||||||||||
ских |
данных |
химические |
вещества |
лело более 10 млн человек, а количе |
|||||||||||||||||||
или другие факторы, на которые име- |
ство умерших составило 8 млн. По ко- |
||||||||||||||||||||||
3 - 7908 Д. Г. Заридзе |
33 |

Т а б л и ц а 1.1. Смертность от злокачественных опухолей ( 3 0 ) в 45 странах мира (2000 г.)
Страна |
Все 3 0 |
|
ободочной и |
молочной |
|
|
|||
|
1 1 U J 1 U C 111 [} 1 « |
прямой кишки |
железы |
|
|
|
|
|
М |
Ж |
м |
Ж |
М |
Ж |
Ж |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Австралия |
150,9 |
(28) |
103,2 |
(25) |
2,2 |
(27) |
0,9 |
(10) |
20,1 |
(12) |
14,4 |
(12) |
19,7 |
(18) |
Австрия |
168,6 |
(20) |
113,8 |
(12) |
3,7 |
(15) |
0,8 |
(18) |
23,0 |
(8) |
14,9 |
(10) |
23,3 |
(9) |
Азербайджан |
114,2 |
(41) |
61,8 |
(45) |
1,3 |
(41) |
0,5 |
(42) |
6,4 |
(40) |
4,8 |
(42) |
8,8 |
(43) |
Болгария |
150,3 |
(29) |
89,4 |
(35) |
2,9 |
(21) |
0,5 |
(43) |
17,8 |
(20) |
12,0 |
(21) |
16,7 |
(31) |
Великобритания |
171,0 |
(18) |
128,0 |
(4) |
1,8 |
(35) |
0,8 |
(32) |
18,7 |
(15) |
13,8 |
(14) |
26,8 |
(3) |
Венгрия |
272,3 |
(1) |
147,4 |
(1) |
10,9 |
(1) |
1,6 |
(2) |
33,5 |
(2) |
20,9 |
(1) |
25,3 |
(7) |
Венесуэла |
104,1 |
(43) |
91,8 |
(33) |
1,3 |
(44) |
0,9 |
(16) |
5,8 |
(43) |
6,1 |
(40) |
11,6 |
(39) |
Германия |
176,6 |
(16) |
116,9 |
(8) |
3,2 |
(19) |
0,8 |
(23) |
21,7 |
(11) |
17,0 |
(6) |
23,7 |
(8) |
Греция |
149,5 |
(31) |
81,8 |
(42) |
1,5 |
(37) |
0,5 |
(44) |
8,4 |
(37) |
6,7 |
(38) |
16,7 |
(32) |
Дания |
184,9 |
(14) |
144,0 |
(2) |
3,0 |
(20) |
1,2 |
(3) |
23,8 |
(7) |
18,5 |
(4) |
29,2 |
(1) |
Израиль |
135,1 |
(38) |
111,4 |
(15) |
1,3 |
(42) |
0,7 |
(33) |
19,7 |
(13) |
15,3 |
(9) |
26,2 |
(4) |
Ирландия |
170,2 |
(19) |
127,8 |
(5) |
3,4(17) |
0,8 |
(24) |
22,6 |
(9) |
15,4 |
(8) |
25,8 |
(6) |
|
Испания |
176,1 |
(17) |
85,0 |
(40) |
3,9 |
(14) |
0,8 |
(31) |
17,3 |
(23) |
11,1 |
(27) |
18,1 |
(25) |
Казахстан |
201,9 |
(8) |
102,6 |
(27) |
2,5 |
(22) |
1,2 |
(4) |
12,2 |
(31) |
8,6 |
(33) |
13,3 |
(36) |
Канада |
160,5 |
(23) |
116,7 |
(9) |
2,3 |
(25) |
0,8 |
(19) |
16,4 |
(26) |
11,6 |
(23) |
22,7 |
(10) |
Киргизия |
185,6 |
(13) |
112,6 |
(14) |
2,1 |
(31) |
0,7 |
(34) |
10,9 |
(35) |
7,9 |
(35) |
17,0 |
(29) |
Китай |
143,3 |
(33) |
76,9 |
(43) |
2,2 |
(28) |
1,0 |
(6) |
7,2 |
(38) |
5,3 |
(41) |
4,5 |
(45) |
Колумбия |
116,1 |
(40) |
106,5 |
(19) |
1,4 |
(39) |
1,0 |
(7) |
5,8 |
(41) |
6,1 |
(39) |
10,6 |
(40) |
Куба |
141,0 |
(35) |
104,0 |
(23) |
4,0 |
(12) |
1,6 |
(1) |
11,4 |
(32) |
12,4 |
(18) |
15,6 |
(35) |
Латвия |
196,7 |
(11) |
102,8 |
(26) |
4,8 |
(8) |
0,7 |
(35) |
17,9 |
(19) |
13,3 |
(15) |
18,1 |
(24) |
Литва |
195,9 |
(12) |
97,0 |
(31) |
5,0 |
(7) |
0,8 |
(26) |
18,0 |
(18) |
10,7 |
(29) |
19,0 |
(20) |
Маврикий |
79,6 |
(45) |
66,3 |
(44) |
2,2 |
(29) |
0,7 |
(37) |
5,8 |
(42) |
3,9 |
(45) |
9,2 |
(41) |
Македония |
140,1 |
(36) |
85,5 |
(38) |
2,1 |
(32) |
0,7 |
(36) |
11,2 |
(34) |
7,8 |
(36) |
17,2 |
(28) |
Мексика |
112,5 |
(42) |
106,3 |
(20) |
1,4 |
(40) |
0,7 |
(38) |
4,7 |
(44) |
4,6 |
(43) |
12,2 |
(38) |
Молдавия |
157,8 |
(25) |
89,4 |
(36) |
6,7 |
(4) |
0,8 |
(29) |
15,8 |
(28) |
10,6 |
(30) |
18,5 |
(21) |
Нидерланды |
182,0 |
(15) |
120,0 |
(7) |
1,5 |
(38) |
0,8 |
(27) |
19,0 |
(14) |
14,0 |
(13) |
27,8 |
(2) |
Новая Зеландия |
167,2 |
(21) |
131,1 |
(3) |
2,3 |
(26) |
0,9 |
(12) |
25,7 |
(4) |
20,2 |
(2) |
25,9 |
(5) |
Норвегия |
155,7 |
(27) |
113,1 |
(13) |
2,4 |
(24) |
0,9 |
(13) |
22,0 |
(10) |
18,0 |
(5) |
20,7 |
(14) |
Польша |
205,2 |
(6) |
111,4 |
(16) |
3,7(16) |
0,8 |
(28) |
16,6 |
(25) |
11,6 |
(24) |
16,8 |
(30) |
|
Португалия |
157,1 |
(26) |
89,1 |
(37) |
3,9(13) |
0,6 |
(41) |
18,5 |
(16) |
11,3 |
(26) |
18,4 |
(22) |
|
Россия |
211,2 |
(5) |
100,6 |
(29) |
5,3 |
(6) |
0,8 |
(30) |
17,5 |
(22) |
12,7 |
(17) |
16,7 |
(33) |
Румыния |
150,0 |
(30) |
90,0 |
(34) |
4,2(11) |
0,9 |
(14) |
11,4 |
(33) |
8,2 |
(34) |
16,2 |
(34) |
|
Словакия |
217,8 |
(4) |
108,8 |
(17) |
9,5 |
(2) |
1,0 |
(9) |
28,0 |
(3) |
16,1 |
(7) |
18,4 |
(23) |
Словения |
203,1 |
(7) |
115,9 |
(11) |
3,4 |
(18) |
0,7 |
(39) |
25,1 |
(5) |
14,6 |
(11) |
20,3 |
(16) |
США |
161,8 |
(22) |
116,4 |
(10) |
1,8 |
(34) |
0,8 |
(17) |
15,9 |
(27) |
12,0 |
(20) |
21,2 |
(12) |
Тринидад и Тобаго 103,5 (44) |
101,9 (39) 2,5 (23) |
1,1 (5) |
8,5 |
(36) |
9,7 |
(31) |
20,6 |
(15) |
||||||
Туркменистан |
117,7 |
(39) |
85,2 |
(39) |
2,2 |
(30) |
0,9 |
(15) |
4,7 |
(45) |
4,1 |
(44) |
9,2 |
(42) |
Финляндия |
145,8 |
(32) |
92,5 |
(32) |
1,7 |
(36) |
0,9 |
(11) |
12,5 |
(30) |
9,5 |
(32) |
17,9 |
(26) |
Франция |
201,5 |
(10) |
98,0 |
(30) |
4,4 |
(10) |
0,8 |
(22) |
18,3 |
(17) |
12,1 |
(19) |
21,4 |
(11) |
Хорватия |
230,1 |
(2) |
105,4 |
(21) |
7,2 |
(3) |
0,8 |
(20) |
24,8 |
(6) |
13,0 |
(16) |
19,9 |
(17) |
Чехия |
222,2 |
(3) |
127,6 |
(6) |
4,4 |
(9) |
0,8 |
(21) |
34,2 |
(1) |
18,5 |
(3) |
21,0 |
(13) |
Чили |
141,2 |
(34) |
108,7 |
(18) |
1,1 |
(45) |
0,4 |
(45) |
7,0 |
(39) |
7,1 |
(37) |
12,7 |
(37) |
Швеция |
137,9 |
(37) |
104,0 |
(24) |
1,3 |
(43) |
0,7 |
(40) |
14,4 |
(29) |
11,5 |
(25) |
17,5 |
(27) |
Эстония |
201,5 |
(9) |
104,8 |
(22) |
5,3 |
(5) |
1,0 |
(8) |
16,7 |
(24) |
12,0 |
(22) |
19,3 |
(19) |
Япония |
159,5 |
(24) |
83,1 |
(41) |
2,0 |
(33) |
0,8 |
(25) |
17,6 |
(21) |
11,0 |
(28) |
7,7 |
(44) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
34
(показатели на 100 тыс. населения, стандартизованные по возрасту)
|
Опухоли |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
предстатель |
|
|
|
матки |
|
|
|
|
|
Лейкозы |
||||
|
|
|
|
|
|
желудка |
|
|
|
|
|
|||
ной железы |
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
шейки |
тела |
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
М |
м |
Ж |
Ж |
|
Ж |
М |
|
Ж |
|
М |
|
Ж |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
18,0 (17) |
36,2 (31) |
14,0 (10) |
2,4 |
(41) |
1,6 (38) |
6,1 (44) |
3,0 |
(44) |
5,7 |
(14) |
3,8 |
(14) |
|
|
18,9 (12) |
41,8 (25) |
10,8 (16) |
4,7 |
(26) |
2,8 (19) |
14,1 (24) |
8,6 |
(22) |
5,0 (25) |
3,6 |
(18) |
||
|
4,3 (43) |
25,5 (37) |
4,5 (42) |
1,9 |
(44) |
3,9 (10) |
24,7 (8) |
10,5 (10) |
4,0 |
(38) |
2,7 (39) |
|||
|
9,0 (34) |
43,7 (22) |
7,1 (32) |
7,4 |
(15) |
3,2 (14) |
17,8 (20) |
9,0 (20) |
5,2 |
(21) |
3,3 (24) |
|||
|
18,5 (14) |
48,6 (18) |
21,1 (4) |
3,9 |
(31) |
1,7 (37) |
10,1 (32) |
4,8 |
(35) |
4,9 |
(29) |
3,3 (29) |
||
|
17,9 (19) |
86,2 (1) |
20,0 (5) |
7,7 |
(14) |
4,1 (8) |
21,0 (16) |
10,1 (13) |
7,6(1) |
4,9 (1) |
||||
|
18,2 (16) |
19,4 (41) |
9,2 (22) |
15,2 |
(2) |
3,7 (12) |
17,5 (22) |
10,0 (14) |
3,9 |
(40) |
3,2 (35) |
|||
|
18,4 (15) |
46,2 (20) |
9,6 (18) |
4,2 |
(27) |
2,1 (31) |
12,9 (27) |
7,8 |
(23) |
5,7 |
(16) |
3,9 (13) |
||
|
10,7 (33) |
50,0 (16) |
7,4 (29) |
2,2 |
(42) |
1,1 (43) |
8,5 (37) |
4,7 (36) |
6,3 (6) |
3,8 |
(15) |
|||
|
23,1 (4) |
50,0 (15) |
26,7 (2) |
4,1 |
(28) |
2,4 (22) |
7,5 (40) |
3,6 (41) |
5,8 |
(13) |
3,9 |
(10) |
||
|
14,2 (30) |
27,5 (36) |
9,3 (21) |
3,1 |
(36) |
1,8 (36) |
9,3 (35) |
5,6 (32) |
6,5 (5) |
4,5 (2) |
||||
|
21,6 (6) |
38,3 (30) |
17,3 (7) |
3,9 |
(29) |
1,5 (39) |
10,1 (31) |
5,0 (34) |
5,4 |
(19) |
3,3 |
(26) |
||
|
15,0 (28) |
49,4 (17) |
4,2 (45) |
2,7 |
(40) |
2,4 (24) |
12,6 (28) |
6,2 (29) |
5,4 |
(20) |
3,2 |
(34) |
||
|
5,2 (41) |
59,5 (9) |
8,3 (25) |
8,1 |
(12) |
2,4 (23) |
32,0 (3) |
13,8 (5) |
3,3 |
(43) |
2,5 (42) |
|||
|
17,1 (21) |
50,4 (14) |
25,0 (3) |
2,8 |
(39) |
1,8 |
(35) |
6,4 (43) |
3,2 (43) |
6,2 (8) |
3,9 (8) |
|||
|
6,4 (39) |
40,7 (27) |
7,3 (30) |
11,3 |
(6) |
4,9 (2) |
47,0 (1) |
18,9(1) |
4,1 |
(37) |
3,2 (30) |
|||
|
1,0 (45) |
33,2 (32) |
13,5 (11) |
3,1 |
(35) |
0,4 (44) |
27,0 (6) |
13,0 (6) |
2,8 |
(44) |
2,0 (44) |
|||
|
15,1 (27) |
17,0 (43) |
8,5 (24) |
13,7 |
(4) |
3,5 (13) |
26,4 (7) |
16,4 (2) |
4,7 |
(31) |
3,9 (9) |
|||
|
22,1 (5) |
42,8 (23) |
15,6 (8) |
10,6 |
(9) |
4,0 (9) |
8,4 (38) |
4,3 (38) |
4,8 |
(30) |
3,6 |
(19) |
||
|
13,0 (31) |
59,1 (10) |
6,3 (37) |
6,6 |
(17) |
4,3 (6) |
24,4 (10) |
10,4 (12) |
6,0 |
(10) |
4,0 (6) |
|||
|
15,6 (24) |
56,5 (11) |
5,5 (39) |
8,8 |
(И) |
3,9 (11) |
24,5 (9) |
9,5 (17) |
5,7 |
(17) |
3,8 |
(16) |
||
|
7,3 (36) |
16,7 (44) |
4,2 (44) |
13,6 |
(5) |
0,2 (45) |
10,6 (29) |
5,7 (30) |
3,4 |
(41) |
2,0 (45) |
|||
|
6,8 (37) |
39,8 (28) |
6,6 (36) |
6,3 |
'18) |
3,0 |
(15) |
21,9 (13) |
9,5 (18) |
4,3 (35) |
2,7 |
(40) |
||
|
16,6 (22) |
22,1 (39) |
8,2 (26) |
17,1 |
(1) |
4,5 (3) |
13,2 (26) |
9,8 |
(15) |
4,9 |
(27) |
4,0 (7) |
||
|
5,0 (42) |
42,1 (24) |
6,2 (38) |
7,0 |
16) |
2,2 (28) |
20,4 (17) |
9,0 |
(21) |
5,2 |
(22) |
3,3 |
(27) |
|
|
20,0 (8) |
59,7 (8) |
14,8 (9) |
2,2 |
43) |
2,2 |
(26) |
9,4 (34) |
4,6 |
(37) |
4,9 |
(28) |
3,2 |
(31) |
|
21,2 (7) |
39,3 (29) |
18,7 (6) |
3,9 |
30) |
2,2 |
(27) |
6,8 (42) |
4,0 |
(39) |
6,3 (7) |
4,4 (4) |
||
|
26,8 (3) |
31,7 (34) |
12,8 (12) |
3,3 |
34) |
3,0 (16) |
9,6 (33) |
5,5 |
(33) |
4,6 (33) |
3,2 (32) |
|||
|
11,2 (32) |
71,5 (2) |
11,3 (15) |
7,8 |
13) |
2,9 |
(18) |
19,2 (19) |
7,3 |
(25) |
5,6 (18) |
3,5 (21) |
||
|
17,9 (20) |
29,5 (35) |
4,8 (40) |
4,8 |
25) |
2,3 |
(25) |
22,2 (12) |
10,9 (8) |
5,1 |
(23) |
3,4 (22) |
||
• |
6,8 (38) |
68,2 (4) |
6,8 (34) |
5,2 |
24) |
2,6 |
(20) |
35,6 (2) |
15,2 (3) |
5,0 |
(26) |
3,4 |
(23) |
|
|
8,3 (35) |
45,1 (21) |
7,3 (31) |
10,9 |
(7) |
2,2 |
(29) |
17,6 (21) |
7,0 |
(27) |
4,5 |
(34) |
3,0 |
(38) |
|
14,3 (29) |
60,7 (7) |
7,8 (27) |
5,4 |
23) |
5,2 (1) |
16,9 (23) |
7,3 (26) |
7,1 (2) |
3,7 |
(17) |
|||
|
18,8 (13) |
55,3 (12) |
10,1 (17) |
5,6 |
22) |
4,4 (5) |
20,2 (18) |
9,6 |
(16) |
5,9 |
(11) |
3,2 |
(33) |
|
|
17,9 (18) |
53,2 (13) |
27,2 (1) |
3,3 |
33) |
2,0 |
(32) |
4,5 (45) |
2,3 |
(45) |
6,6 (4) |
4,2 (5) |
||
|
32,3 (1) |
13,2 (45) |
4,3 (43) |
15,0 |
(3) |
4,3 (7) |
8,7 (36) |
6,9 |
(28) |
3,4 |
(42) |
3,1 |
(36) |
|
|
1,8 (44) |
18,9 (42) |
4,6 (41) |
6,3 |
19) |
1,4 (41) |
21,1 (15) |
10,8 (9) |
2,6 |
(45) |
2,4 |
(43) |
||
|
19,1 (11) |
41,2 (26) |
7,4 (28) |
1,3 |
45) |
2,5 |
(21) |
10,3 (30) |
5,6 |
(31) |
4,7 |
(32) |
3,3 |
(25) |
|
19,2 (10) |
48,5 (19) |
6,7 (35) |
3,5 |
32) |
2,1 |
(30) |
8,0 (39) |
3,6 (42) |
6,1 (9) |
3,9 (12) |
|||
|
15,3 (25) |
70,3 (3) |
9,4 (20) |
5,7 |
21) |
1,9 (34) |
21,7 (14) |
9,1 (19) |
5,8 |
(12) |
3,5 |
(20) |
||
|
15,7 (23) |
65,3 (5) |
11,5 (14) |
6,2 |
20) |
4,4 (4) |
13,5 (25) |
7,5 |
(24) |
6,7 (3) |
4,4 (3) |
|||
|
19,9 (9) |
20,3 (40) |
7,0 (33) |
10,6 |
(8) |
1,4 (40) |
30,1 (5) |
12,7 (7) |
4,0 (39) |
3,0 |
(37) |
|||
|
27,3 (2) |
22,6 (38) |
12,6 (13) |
2,9 |
38) |
2,0 |
(33) |
7,4 (41) |
4,0 |
(40) |
5,1 |
(24) |
3,3 |
(28) |
|
15,3 (26) |
64,5 (6) |
8,6 (23) |
9,7 |
Ю) |
2,9 |
(17) |
24,2 ( И ) |
10,4 (11) |
5,7 (15) |
3,9(11) |
|||
|
5,5 (40) |
33,1 (33) |
9,6 (19) |
3,0 |
37) |
1,2 (42) |
31,2 (4) |
13,8 (4) |
4,1 |
(36) |
2,6 |
(41) |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3* |
35 |
реса. В большинстве эпидемиологиче ских исследований, опубликованных после 1985 г., эти факторы были учте ны. При подсчете ОР делалась по правка (adjustment) на возраст при первом половом контакте, количество сексуальных партнеров, применение барьерных методов контрацепции, на
личие венерических заболеваний |
и |
|
т. д. В |
литературе опубликованы |
ре |
зультаты |
эпидемиологических иссле |
|
дований, в которых изучалась связь между применением оральных кон трацептивов и разными стадиями про цесса канцерогенеза в шейке матки: дисплазией, интраэпителиальным (in situ) и инвазивным раком. Наиболее
убедительные результаты |
получены |
|
для |
интраэпителиального |
рака. Во |
всех |
эпидемиологических |
исследова |
ниях выявлено повышение риска ин траэпителиального рака у женщин, применявших оральные контрацепти вы, а в большинстве из них величина ОР зависела от длительности их при менения. У женщин, применявших эти препараты более 10 лет, ОР был более 2,0, а в исследовании, прове денном в Англии, равен 4,0. Результа ты большинства исследований, в ко торых изучалась связь между исполь зованием оральных контрацептивов и инвазивным раком, также указывают на наличие причинной связи. Необхо димо отметить, что у женщин, приме няющих оральные контрацептивы, повышен риск и аденокарциномы цервикального канала — опухоли, за болеваемость которой в отличие от плоскоклеточного рака шейки матки имеет тенденцию роста у молодых женщин.
Длительное применение ораль ных контрацептивов скорее всего по вышает риск печеночно-клеточного рака и аденомы печени, а также рака щитовидной железы и меланомы кожи.
Гормонозаместительная терапия.
Гормонозаместительная терапия полу чила широкое распространение в США в 70-х годах XX в. Однако выяс нилось, что женщины, получавшие
это лечение, часто заболевали раком тела матки. В 80-х годах на смену пре паратам, содержащим только эстроге ны, пришли так называемые цикличе ские препараты, которые содержат эс трогены и прогестерон. В настоящее время интерес с точки зрения возмож ного канцерогенного риска представ ляют именно они. Добавление прогес терона к эстрогенсодержащим препа ратам привело к снижению связанной с эстрогенами митотической активно сти эндометрия и соответственно рис ку рака эндометрия. Тем не менее у женщин, получающих в течение дли тельного времени (более 8—10 лет) современную гормонозаместительную терапию, отмечается небольшое повышение риска рака эндометрия. В связи с этим продолжаются исследо вания в области создания препаратов с оптимальными дозами эстрогенов и прогестинов, которые снижали бы риск остеопороза и сердечно-сосуди стых заболеваний и в то же время не вызывали повышения онкологическо го риска.
Гормонозаместительная терапия повышает ОР рака молочной железы на 35 %. Однако через 5 лет после за вершения приема этих препаратов ОР рака молочной железы снижается до нормального. На основании анализа результатов нескольких эпидемиоло гических исследований, посвященных этой теме, подсчитано, что у женщин в возрасте 50—60 лет, получающих это лечение, кумулятивный риск рака мо лочной железы составляет 6 дополни тельных случаев на 1 тыс. населения, т. е. среди 1 тыс. женщин, не получав ших гормонозаместительную терапию, раком молочной железы заболеют 46, а среди 1 тыс. женщин, которые полу чали это лечение, — 51. Что же каса ется влияния применения гормонозаместительной терапии на риск рака яичников, то данные по этому вопро су противоречивы. В ряде ранних ра бот было выявлено небольшое повы шение риска рака яичников, связан ного с приемом эстрогенсодержащих препаратов. Данных, указывающих на
58
Т а б л и ц а 1.2. Стандартизованные показатели заболеваемости злокачественными новообра-
|
|
|
Полости рта |
Пищевод |
Желудок |
Толстая |
||||
|
|
|
и глотки |
кишка |
||||||
|
|
Страна |
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
М |
Ж |
М |
Ж |
М |
Ж |
М |
Ж |
|
|
|
|
|
|
|
|
|
|
|
АЗИЯ |
|
|
|
|
|
|
|
|
|
|
Китай, |
Шанхай |
7,2 |
3,7 |
12,5 |
4,8 |
46,5 |
21,0 |
12,2 |
10,8 |
|
Гонконг |
|
31,7 |
12,6 |
14,2 |
3,2 |
19,4 |
9,5 |
22,5 |
18,8 |
|
Израиль, евреи |
7,2 |
ЗД |
1,7 |
1,1 |
13,0 |
6,2 |
24,9 |
19,9 |
||
не |
евреи |
3,5 |
2,3 |
0,5 |
0,2 |
6,8 |
3,2 |
6,2 |
6,1 |
|
Япония, Осака |
6,0 |
2,1 |
9,1 |
1,6 |
65,0 |
27,3 |
20,7 |
13,1 |
||
АМЕРИКА |
|
|
|
|
|
|
|
|
||
Бразилия, |
Порт Алегре |
18,2 |
2,6 |
18,9 |
4,1 |
27,9 |
8,9 |
15,7 |
13,7 |
|
Канада |
|
|
13,0 |
4,1 |
4,1 |
1,3 |
10,6 |
4,5 |
26,9 |
21,3 |
Колумбия, |
Кали |
6,2 |
3,5 |
4,0 |
2,3 |
33,3 |
19,3 |
6,5 |
6,3 |
|
США, |
белые |
12,2 |
5,0 |
4,5 |
1,3 |
7,5 |
3,1 |
28,1 |
20,8 |
|
США, |
черные |
19,5 |
5,8 |
13,8 |
3,9 |
14,5 |
5,9 |
33,6 |
26,9 |
|
АФРИКА |
|
|
|
|
|
|
|
|
|
|
Зимбабве, |
черные |
5,1 |
2,7 |
30,4 |
8,0 |
13,8 |
18,4 |
6,6 |
3,0 |
|
Зимбабве, |
белые |
6,4 |
10,7 |
2,4 |
1,5 |
8,3 |
6,3 |
17,8 |
14,1 |
|
ЕВРОПА |
|
|
|
|
|
|
|
|
|
|
Англия |
и Уэльс |
5,7 |
2,5 |
7,6 |
3,5 |
16,1 |
6,2 |
19,3 |
15,6 |
|
Беларусь |
|
17,6 |
2,3 |
5,8 |
0,6 |
46,8 |
20,1 |
9,8 |
7,6 |
|
Германия, |
Саар |
18,7 |
3,3 |
6,7 |
0,8 |
18,5 |
9,0 |
25,5 |
20,4 |
|
восточные земли |
8,8 |
1,7 |
4,1 |
0,6 |
20,1 |
10,5 |
15,5 |
13,9 |
||
Дания |
|
|
9,9 |
3,6 |
4,8 |
1,4 |
9,0 |
4,7 |
20,6 |
19,9 |
Испания, Сарагоса |
16,0 |
1,3 |
5,5 |
0,5 |
19,6 |
8,4 |
13,0 |
10,2 |
||
Италия, Варезе |
16,0 |
2,3 |
6,8 |
0,7 |
26,6 |
12,7 |
27,4 |
18,7 |
||
Нидерланды |
8,7 |
3,5 |
5,2 |
1,9 |
15,4 |
6,1 |
21,9 |
18,9 |
||
Польша, Варшава |
9,6 |
2,4 |
4,8 |
1,1 |
19,1 |
6,9 |
16,0 |
10,0 |
||
Финляндия |
7,5 |
2,8 |
3,5 |
1,7 |
16,6 |
9,2 |
12,8 |
11,6 |
||
Франция, Изер |
24,8 |
3,6 |
10,0 |
1,0 |
11,8 |
4,7 |
22,3 |
15,5 |
||
Чехия |
|
|
10,8 |
2,2 |
4,2 |
0,5 |
19,5 |
9,2 |
24,0 |
15,7 |
Швейцария, Женева |
22,0 |
6,4 |
7,2 |
1,4 |
12,3 |
5,5 |
25,2 |
15,4 |
||
Швеция |
|
6,8 |
3,0 |
3,0 |
1,0 |
10,7 |
5,4 |
17,7 |
15,9 |
|
Шотландия |
9,4 |
3,7 |
9,4 |
5,0 |
17,6 |
7,5 |
23,7 |
19,4 |
||
Эстония |
|
15,1 |
2,0 |
5,6 |
0,7 |
34,0 |
16,6 |
14,1 |
11,4 |
|
ОКЕАНИЯ |
|
|
|
|
|
|
|
|
||
Австралия, |
Виктория |
15,4 |
4,9 |
5,4 |
2,4 |
11,7 |
4,9 |
27,5 |
22,9 |
|
Гаваи, |
белые |
16,7 |
6,0 |
4,4 |
2,0 |
7,8 |
4,3 |
32,7 |
22,9 |
|
Гаваи, |
японцы |
8,2 |
2,2 |
3,7 |
0,6 |
21,5 |
10,7 |
34,4 |
22,6 |
|
Гаваи, |
гавайцы |
12,0 |
3,3 |
7,6 |
1,3 |
15,1 |
10,4 |
19,9 |
16,6 |
|
Гаваи, |
китайцы |
10,0 |
7,2 |
4,5 |
0,0 |
12,3 |
5,2 |
22,5 |
18,1 |
|
Новая Зеландия, не маори |
10,5 |
4,0 |
5,3 |
2,4 |
11,1 |
4,8 |
31,2 |
29,6 |
||
маори |
|
11,4 |
4,1 |
9,2 |
2,1 |
28,0 |
13,8 |
21,5 |
16,0 |
|
|
|
|
|
|
|
|
|
|
|
|
личеству как заболевших, так и умер ших на первом месте стоит рак легко го, которым в 2000 г. заболели 1 млн 230 тыс. человек и умерли 1 млн 100 тыс. Второе место в структуре заболе
ваемости и смертности от злокачест венных опухолей в мире занимает рак желудка. По количеству заболевших на третьем месте стоит рак молочной железы. Однако по числу умерших рак
36
зованиями на 100 тыс. населения (Cancer Incidence in five continents, vol. VII, IARC, 1997)
Прямая кишка |
Печень |
Поджелудоч |
Гортань |
Легкое |
||||||
ная железа |
||||||||||
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
M |
Ж |
М |
Ж |
М |
Ж |
М |
Ж |
М |
Ж |
|
|
|
|
|
|
|
|
|
|
|
|
9,3 |
7,3 |
28,2 |
9,8 |
6,3 |
4,1 |
3,3 |
0,4 |
56,1 |
18,2 |
|
12,6 |
9,2 |
36,2 |
9,5 |
4,0 |
2,9 |
7,7 |
0,8 |
74,7 |
30,7 |
|
14,4 |
11,4 |
3,2 |
1,6 |
7,2 |
5,7 |
4,7 |
0,7 |
27,0 |
9,2 |
|
3,1 |
3,2 |
2,6 |
1,4 |
4,0 |
2,9 |
3,7 |
0,0 |
29,1 |
3,7 |
|
13,5 |
6,8 |
46,7 |
11,5 |
9,6 |
5,3 |
3,4 |
0,3 |
43,5 |
12,4 |
|
12,4 |
6,8 |
8,3 |
3,5 |
9,0 |
6,3 |
10,5 |
1,3 |
67,5 |
14,4 |
|
16,1 |
9,2 |
3,1 |
1,2 |
7,7 |
5,5 |
6,2 |
1,2 |
65,4 |
28,0 |
|
5,4 |
4,5 |
2,6 |
2,2 |
5,4 |
3,8 |
5,2 |
1,1 |
24,4 |
9,5 |
|
14,3 |
8,7 |
3,0 |
1,2 |
7,7 |
5,7 |
6,3 |
1,4 |
61,3 |
33,8 |
|
12,8 |
8,5 |
6,5 |
2,0 |
11,8 |
9,3 |
11,0 |
2,4 |
99,1 |
38,4 |
|
3,8 |
2,6 |
34,6 |
19,2 |
5,9 |
8,0 |
4,5 |
1,0 |
24,9 |
7,3 |
|
22,4 |
7,4 |
12,9 |
4,7 |
4,2 |
6,1 |
9,9 |
0,0 |
36,0 |
18,1 |
|
14,6 |
8,1 |
2,0 |
1,0 |
7,4 |
5,3 |
4,5 |
0,8 |
62,4 |
22,8 |
|
12,3 |
8,3 |
4,1 |
1,9 |
8,6 |
3,6 |
10,5 |
0,3 |
66,8 |
6,0 |
|
17,3 |
10,9 |
4,2 |
1,6 |
6,9 |
4,2 |
8,1 |
0,7 |
70,9 |
10,3 |
|
15,7 |
10,1 |
5Д |
2,3 |
7,3 |
4,5 |
5,1 |
0,3 |
59,2 |
7,6 |
|
17,0 |
10,4 |
3,7 |
2,0 |
7,8 |
6,4 |
5,5 |
1,1 |
51,9 |
25,4 |
|
11,1 |
6,4 |
5,5 |
2,3 |
5,9 |
3,4 |
17,1 |
0,1 |
48,0 |
2,7 |
|
16,1 |
8,2 |
13,0 |
3,3 |
8,8 |
5,7 |
12,7 |
0,6 |
77,6 |
8,5 |
|
14,5 |
8,9 |
1,6 |
0,6 |
6,8 |
4,7 |
6,3 |
0,8 |
73,0 |
13,0 |
|
11,3 |
6,8 |
5,0 |
3,0 |
7,9 |
5,0 |
10,9 |
1,2 |
68,4 |
19,5 |
|
10,5 |
6,6 |
4,8 |
2,7 |
9,5 |
7,0 |
3,3 |
0,3 |
54,3 |
8,2 |
|
15,4 |
9,8 |
9,1 |
0,9 |
4,6 |
2,8 |
9,8 |
0,5 |
50,1 |
5,6 |
|
24,2 |
11,6 |
6,6 |
2,7 |
11,4 |
6,5 |
7,4 |
0,4 |
77,9 |
10,1 |
|
12,3 |
7,0 |
11,2 |
1,6 |
9,2 |
6,3 |
8,6 |
1,0 |
53,7 |
14,3 |
|
12,1 |
8,3 |
3,4 |
1,8 |
7,0 |
5,7 |
2,5 |
0,4 |
23,9 |
10,9 |
|
14,3 |
8,3 |
3,1 |
1,4 |
8,1 |
5,5 |
6,2 |
1,3 |
79,8 |
33,8 |
|
11,6 |
7,5 |
4,2 |
1,9 |
10,7 |
5,7 |
9,1 |
0,4 |
75,7 |
8,3 |
|
|
||||||||||
19,2 |
11,0 |
3,0 |
0,7 |
6,8 |
4,4 |
4,8 |
0,6 |
46,0 |
15,8 |
|
14,0 |
8,0 |
4,5 |
1,5 |
8,5 |
5,7 |
8,3 |
1,2 |
59,6 |
37,9 |
|
19,0 |
7,9 |
5,0 |
1,9 |
5,8 |
5,8 |
2,2 |
0,2 |
34,0 |
11,1 |
|
13,9 |
8,2 |
6,8 |
2,6 |
8,8 |
7,2 |
3,8 |
0,6 |
72,3 |
35,0 |
|
12,1 |
6,4 |
10,5 |
3,2 |
5,8 |
4,0 |
2,0 |
0,0 |
37,6 |
19,0 |
|
20,1 |
11,2 |
2,9 |
1,4 |
7,3 |
4,6 |
4,0 |
0,6 |
46,5 |
18,2 |
|
12,8 |
9,2 |
12,8 |
3,6 |
9,8 |
6,7 |
3,1 |
1,2 |
99,7 |
72,9 |
|
|
|
|
|
|
|
|
|
|
|
|
молочной железы стоит на пятом мес |
(0,79), что говорит о значительно луч |
те. Соотношение количества умерших |
шем прогнозе для этого заболевания. |
и заболевших для рака молочной же |
По числу заболевших на четвертом и |
лезы существенно ниже (0,39), чем |
пятом местах стоят рак ободочной и |
для рака легкого (0,89) и желудка |
прямой кишки и рак печени, по коли- |
37
|
|
|
Меланома |
Про |
Мочевой пу |
Лимфогрануле |
|||||
|
|
|
стата |
|
зырь |
|
матоз |
||||
|
|
Страна |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
М |
Ж |
М |
М |
|
Ж |
М |
|
Ж |
|
|
|
|
|
|
|
|
|
|
|
|
АЗИЯ |
|
|
|
|
|
|
|
|
|
|
|
Гонконг |
|
1,0 |
0,8 |
7,9 |
14,5 |
|
4,3 |
0,7 |
|
0,3 |
|
Израиль, евреи |
9,6 |
9,8 |
23,9 |
25,2 |
|
5,3 |
2,9 |
|
2,8 |
||
не |
евреи |
0,7 |
0,7 |
10,4 |
13,1 |
|
1,4 |
2,4 |
|
1,6 |
|
Китай, |
Шанхай |
0,3 |
0,3 |
2,3 |
6,9 |
|
1,8 |
0,4 |
|
0,3 |
|
Япония, Осака |
0,2 |
0,2 |
6,8 |
7,4 |
|
1,8 |
0,5 |
|
0,2 |
||
АМЕРИКА |
|
|
|
|
|
|
|
|
|
||
Бразилия, |
Порт Алегре |
5,2 |
5,0 |
42,8 |
18,6 |
|
3,5 |
2,5 |
|
1,1 |
|
Канада |
|
7,7 |
6,9 |
64,7 |
18,7 |
|
5,0 |
2,9 |
|
2,3 |
|
Колумбия, |
Кали |
2,7 |
3,1 |
32,7 |
7,4 |
|
2,7 |
1,9 |
|
0,9 |
|
США, |
белые |
13,1 |
10,2 |
100,8 |
24,1 |
|
6,2 |
3,4 |
|
2,7 |
|
США, |
черные |
0,8 |
0,5 |
137,0 |
11,1 |
|
4,3 |
2,5 |
|
2,0 |
|
АФРИКА |
|
|
|
|
|
|
|
|
|
|
|
Зимбабве, |
черные |
1,8 |
3,8 |
29,2 |
13,2 |
|
12,5 |
1,0 |
|
0,6 |
|
Зимбабве, |
белые |
18,6 |
15,0 |
55,7 |
27,4 |
|
7,8 |
0,0 |
|
4,3 |
|
ЕВРОПА |
|
|
|
|
|
|
|
|
|
|
|
Англия |
и Уэльс |
4,6 |
6,5 |
28,0 |
20,3 |
|
5,7 |
2,5 |
|
1,8 |
|
Беларусь |
|
1,9 |
2,3 |
12,2 |
12,4 |
|
1,4 |
3,3 |
|
2,5 |
|
Германия, |
Саар |
5,8 |
6,1 |
35,9 |
23,1 |
|
5,2 |
2,3 |
|
1,9 |
|
восточные земли |
5,0 |
5,3 |
23,7 |
19,1 |
|
3,5 |
2,6 |
|
2,0 |
||
Дания |
|
|
8,8 |
11,7 |
31,0 |
27,9 |
|
7,7 |
2,9 |
|
1,6 |
Испания, Сарагоса |
2,2 |
3,3 |
19,7 |
22,4 |
|
2,6 |
2,2 |
|
1,7 |
||
Италия, Варезе |
5,1 |
5,0 |
28,2 |
35,0 |
|
5,1 |
3,5 |
|
2,9 |
||
Нидерланды |
6,9 |
9,8 |
39,6 |
15,2 |
|
3,0 |
2,4 |
|
1,7 |
||
Польша, Варшава |
3,8 |
3,7 |
15,8 |
13,1 |
|
3,0 |
2,1 |
|
1,7 |
||
Финляндия |
7,8 |
6,7 |
41,3 |
15,2 |
|
3,0 |
2,4 |
|
1,8 |
||
Франция, Изер |
4,1 |
6,2 |
48,0 |
16,0 |
|
2,6 |
2,4 |
|
1,2 |
||
Чехия |
|
|
6,5 |
6,1 |
24,1 |
15,4 |
|
3,2 |
3,0 |
|
2,4 |
Швейцария, Женева |
10,5 |
11,1 |
49,0 |
32,5 |
|
6,3 |
2,7 |
|
1,7 |
||
Швеция |
|
11,0 |
11,1 |
55,3 |
17,4 |
|
4,6 |
2,2 |
|
1,6 |
|
Шотландия |
6,0 |
8,3 |
31,2 |
22,9 |
|
7,3 |
2,6 |
|
1,9 |
||
Эстония |
|
3,6 |
4,1 |
21,6 |
11,2 |
|
2,0 |
2,7 |
|
1,6 |
|
ОКЕАНИЯ |
|
|
|
|
|
|
|
|
|
||
Австралия, |
Виктория |
22,4 |
20,3 |
47,5 |
18,3 |
|
5,0 |
2,3 |
|
1,7 |
|
Гаваи, |
белые |
19,5 |
12,4 |
108,2 |
24,2 |
|
5,9 |
2,7 |
|
2,9 |
|
Гаваи, |
японцы |
0,3 |
0,3 |
64,2 |
10,9 |
|
3,3 |
0,8 |
|
1,3 |
|
Гаваи, |
гавайцы |
0,9 |
1,2 |
42,1 |
3,9 |
|
3,2 |
0,8 |
|
2,0 |
|
Гаваи, |
китайцы |
0,8 |
0,7 |
62,9 |
9,6 |
|
2,7 |
0,5 |
|
0,1 |
|
Новая Зеландия, не маори |
25,0 |
29,8 |
37,8 |
14,0 |
|
4,1 |
1,9 |
|
1,3 |
||
маори |
|
5,1 |
3,4 |
44,4 |
10,5 |
|
2,2 |
1,6 |
|
0,6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
честву же умерших рак ободочной и |
Рак легкого. Раком легкого значи- |
|
прямой кишки и рак печени занима- |
тельно чаще болеют мужчины, чем |
|
ют третье и четвертое места. Ниже |
женщины. По данным МАИР, очет> |
|
представлены данные дескриптивной |
высокая заболеваемость раком легкого |
|
эпидемиологии 15 наиболее частых |
зарегистрирована |
среди чернокожего |
форм злокачественных опухолей. |
населения США. |
Заболеваемость так |
38
|
|
|
|
|
|
|
|
|
Продолжение |
|||
|
|
|
|
|
|
|
|
|
|
|
||
Лимфома |
Лейкемия |
Молочная |
|
Шейка |
Тело |
|
Яичники |
|||||
железа |
|
матки |
матки |
|
||||||||
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
М |
Ж |
М |
Ж |
Ж |
|
Ж |
|
Ж |
|
Ж |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
8,7 |
6,2 |
5,9 |
4,2 |
34,0 |
|
15,3 |
|
7,0 |
|
7,4 |
|
|
12,6 |
10,4 |
6,5 |
4,4 |
77,4 |
|
5,3 |
|
10,8 |
|
11,6 |
|
|
8,3 |
6,5 |
4,7 |
3,3 |
21,3 |
|
3,0 |
|
4,9 |
|
3,0 |
|
|
4,3 |
2,5 |
3,8 |
2,8 |
26,5 |
|
3,3 |
|
3,7 |
|
5,8 |
|
|
6,1 |
3,6 |
5,0 |
3,4 |
24,3 |
|
9,2 |
|
3,0 |
|
5,6 |
|
|
8,4 |
6,4 |
6,1 |
4,8 |
62,0 |
|
22,1 |
|
6,2 |
|
8,4 |
|
|
13,1 |
9,1 |
9,7 |
6,1 |
76,8 |
|
7,8 |
|
14,3 |
|
10,5 |
|
|
7,9 |
6,0 |
4,2 |
4,1 |
38,8 |
|
34,4 |
|
6,5 |
|
8,9 |
|
|
16,3 |
10,1 |
9,9 |
6,1 |
90,7 |
|
7,5 |
|
18,2 |
|
П,9 |
||
12,3 |
7,0 |
8,2 |
5,5 |
79,3 |
|
12,0 |
|
11,4 |
|
8,1 |
|
|
4,7 |
4,0 |
5,2 |
4,7 |
20,4 |
|
67,2 |
|
4,3 |
|
8,0 |
|
|
4,1 |
3,9 |
6,1 |
3,4 |
127,7 |
|
10,4 |
|
9,9 |
|
14,1 |
|
|
9,6 |
6,3 |
7,8 |
5,0 |
68,8 |
|
12,5 |
|
8,7 |
|
12,4 |
|
|
3,2 |
1,8 |
8,7 |
5,0 |
29,6 |
|
11,2 |
|
10,6 |
|
10,7 |
|
|
9,4 |
5,8 |
8,7 |
5,2 |
61,5 |
|
11,4 |
|
12,7 |
|
9,6 |
|
|
5,6 |
3,7 |
6,4 |
4,2 |
48,2 |
|
21,3 |
|
13,9 |
|
12,2 |
|
|
9,8 |
6,5 |
9,3 |
5,8 |
73,2 |
|
15,2 |
|
14,7 |
|
14,0 |
|
|
7,0 |
5,2 |
5,9 |
5,2 |
40,4 |
|
4,8 |
|
8,7 |
|
8,1 |
|
|
14,0 |
10,1 |
9,7 |
6,3 |
73,5 |
|
6,4 |
|
12,6 |
|
10,2 |
|
|
10,6 |
7,0 |
7,3 |
4,5 |
79,6 |
|
7,2 |
|
10,8 |
|
11,2 |
|
|
6,4 |
3,2 |
6,0 |
4,3 |
43,2 |
|
15,2 |
|
13,0 |
|
13,3 |
|
|
10,5 |
7,8 |
6,9 |
4,9 |
65,0 |
|
3,6 |
|
12,9 |
|
10,9 |
|
|
10,6 |
6,9 |
8,0 |
4,2 |
86,0 |
|
9,7 |
|
10,0 |
|
9,1 |
|
|
7,0 |
4,4 |
8,7 |
5,4 |
45,1 |
|
16,4 |
|
17,7 |
|
13,3 |
|
|
13,5 |
8,1 |
10,4 |
6,0 |
77,8 |
|
6,1 |
|
12,7 |
|
11,9 |
|
|
10,8 |
6,9 |
7,5 |
5,2 |
72,9 |
|
8,0 |
|
13,2 |
|
13,2 |
|
|
9,4 |
7,4 |
8,1 |
5,2 |
72,7 |
|
12,7 |
|
7,4 |
|
13,3 |
|
|
4,5 |
2,5 |
8,0 |
4,8 |
36,5 |
|
14,1 |
|
12,9 |
|
12,5 |
|
|
13,8 |
9,1 |
9,5 |
5,5 |
66,7 |
|
9,4 |
|
10,2 |
|
9,6 |
|
|
15,1 |
6,9 |
9,6 |
4,6 |
96,5 |
|
10,5 |
|
15,7 |
|
14,2 |
|
|
8,5 |
6,3 |
4,4 |
4,1 |
72,9 |
|
6,4 |
|
14,2 |
|
8,1 |
|
|
11,0 |
5,0 |
8,4 |
6,4 |
83,9 |
|
8,6 |
|
20,6 |
|
8,3 |
|
|
12,8 |
8,7 |
5,2 |
1,4 |
57,6 |
|
4,5 |
|
14,0 |
|
6,6 |
|
|
10,3 |
7,5 |
10,1 |
6,6 |
77,2 |
|
11,9 |
|
9,4 |
|
11,0 |
|
|
8,1 |
6,7 |
10,7 |
6,4 |
77,1 |
|
32,2 |
|
15,8 |
|
12,2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
же высока среди "белого" населения |
ности от |
рака |
легкого |
среди |
мужчин |
|||||||
Новой Зеландии, в странах Восточной |
отмечаются в |
странах |
Восточной Ев- |
|||||||||
иЗападной Европы. Относительно ропы (Венгрия, Польша, Хорватия,
низкие показатели отмечены в Китае, |
Россия), а также в странах Западной |
Японии и в африканских регистрах. |
Европы и Северной Америки. Самая |
Самые высокие показатели смерт- |
высокая смертность среди женщин за- |
39
регистрирована в США, Дании и Ка наде. Относительно низкие показате ли смертности зафиксированы в стра нах Азии и Южной Америки.
Заболеваемость и смертность от рака легкого растет во всем мире, за исключением США и некоторых стран Северной и Западной Европы, где наметилось снижение заболевае мости среди мужчин. Рост частоты ра ка легкого более выражен у женщин, чем у мужчин.
Рак легкого продолжает оставаться заболеванием с очень неблагоприят ным прогнозом. Средняя пятилетняя выживаемость в странах Европы равна 8 %, а в США - 14 %.
Рак желудка. Заболеваемость ра ком желудка значительно выше у мужчин, чем женщин. Высокая забо леваемость зарегистрирована в Япо нии, Китае, в странах Восточной Ев ропы и Южной Америки. Самые низ кие показатели фиксируются в США и некоторых западноевропейских странах.
Самая высокая смертность от рака желудка в России, Казахстане, Чили и Японии, а также в большинстве стран Восточной Европы. Самые низкие по казатели смертности в США, Канаде, Новой Зеландии, странах Западной и Северной Европы.
Заболеваемость раком желудка снижается у мигрантов из стран с вы сокой заболеваемостью в страны, где она низка. Наиболее характерным примером этого феномена является снижение заболеваемости раком же лудка у японских и китайских пересе ленцев в США, что подтверждает роль факторов образа жизни, в частности питания, в этиологии этого заболева ния.
В целом в мире, как и в отдельных странах, отмечается снижение заболе ваемости и смертности от рака желуд ка. Стандартизованная по возрасту за болеваемость раком желудка в мире ежегодно снижается на 4—5 %.
Средняя пятилетняя выживаемость при заболевании раком желудка в За падной Европе равна 18 %. Самый
высокий показатель пятилетней вы живаемости (53 %) зафиксирован в Японии, и его можно объяснить про ведением в этой стране массового скрининга. Пятилетняя выживаемость в мире колеблется в пределах 10— 20 %.
Рак молочной железы. Заболевае мость раком молочной железы высока практически во всех развитых странах мира, кроме Японии, и низка в стра нах Азии, Африки и Южной Америки. Самая высокая смертность от рака молочной железы зафиксирована в Дании, Нидерландах, Великобрита нии, Израиле.
Заболеваемость раком молочной железы растет у мигрантов из стран, где она низкая, в страны с высокой заболеваемостью. Так, например, за болеваемость раком молочной железы значительно выше среди японцев и китайцев, проживающих в США, чем в Японии и Китае.
Заболеваемость раком молочной железы растет в большинстве стран мира, а особенно в странах Восточной Азии. В Китае ежегодный прирост за болеваемости раком молочной железы составил 5 %. В то же время смерт ность от рака молочной железы во многих западных странах начала сни жаться. Например, в США темп сни жения за 1 год составляет 1,7 %.
Прогноз рака молочной железы достаточно благоприятен. Наилуч ший показатель пятилетней выживае мости отмечен в США (84 %), Австра лии (73 %), Японии (74 %). Показа тель выживаемости в Европе состав ляет 63—67 %. В целом же в мире пя тилетняя выживаемость равна 50— 60 %.
Рак ободочной и прямой кишки.
Самая высокая смертность от рака ободочной и прямой кишки зафикси рована в Чехии, Венгрии, Словакии, Новой Зеландии. В целом показатели смертности от рака этой локализации высоки в странах Северной Америки и Европы, низки в странах Азии и особенно Африки.
Заболеваемость раком ободочной
40
кишки одинакова среди мужчин и женщин. Самая высокая заболевае мость раком ободочной кишки заре гистрирована среди "белого" населе ния и выходцев из Японии, живущих на Гавайских островах и в некоторых штатах Канады и США. Заболевае мость раком ободочной кишки низка среди африканского населения в Аф рике и Азии, кроме Японии, где за последние два десятилетия она значи тельно выросла.
Раком прямой кишки чаще болеют мужчины, чем женщины. Самая высо кая заболеваемость зарегистрирована в Чехии. Высокая заболеваемость ре гистрируется в Австралии и Новой Зе ландии, Северной Америке, Северной и Западной Европе. Низкая заболе ваемость в странах Африки и Азии, за исключением Японии, где заболевае мость раком прямой кишки значи тельно выросла и сравнима с показа телями в Европе.
Исследование мигрантов показа ли, что у переселенцев из стран с низкой заболеваемостью раком тол стой кишки в страны с высокой забо леваемостью растет частота рака это го органа. Например, заболеваемость раком ободочной кишки среди япон цев, проживающих в США, значи тельно выше, чем в Японии, и даже выше, чем у "белых" американцев, хотя, как уже говорилось, в Японии зафиксирован рост заболеваемости раком этого органа. Аналогичный тренд отмечен и среди китайских ми грантов в США.
Заболеваемость раком ободочной кишки растет практически во всех странах, кроме США и ряда европей ских стран, где она стабилизирова лась, а в некоторых из них начала снижаться. Аналогичны тенденции в показателях смертности от рака пря мой кишки. В странах с исходной низкой частотой этого заболевания смертность растет; в ряде же стран от мечается стабилизация или снижение показателей смертности. Снижение смертности от рака толстой кишки происходит в Северной Америке, Ав
стралии, Новой Зеландии и Западной Европе.
Прогноз при раке ободочной и прямой кишки относительно благо приятен. Пятилетняя выживаемость в целом в мире колеблется в пределах 30—40 %. Лучший показатель выжи ваемости зафиксирован в США (60 %). В Западной Европе 5 'лет вы живают 41 % больных раком толстой кишки.
Рак печени. Основная часть всех случаев печеночно-клеточного рака (80 %) зарегистрирована в развиваю щихся странах, а именно в Западной и Центральной Африке и Юго-Восточ ной Азии, где доля рака печени со ставляет 25 % и более в структуре смертности от злокачественных опу холей. В Европе (за исключением не которых регионов Южной Европы), Северной Америке и Австралии забо леваемость и смертность от рака пече ни низка. Пятилетняя выживаемость больных раком печени не превышает 3-5 %.
Рак простаты. Рак простаты встре чается в основном у лиц пожилого возраста. В развитых странах 82 % ра ка простаты диагностируется у муж чин старше 65 лет. Самая высокая за болеваемость регистрируется в США среди чернокожих и "белых" мужчин, где рак простаты занимает первое ме сто в структуре заболеваемости злока чественными опухолями мужского на селения. Заболеваемость раком про статы высока в Канаде, странах Се верной и Западной Европы и Австра лии. Высокие показатели заболевае мости отмечены также в Африке сре ди коренного населения.
Самые высокие показатели смерт ности от рака простаты зарегистриро ваны в Тринидаде и Тобаго, Норвегии и Швеции. В США, стране с самой высокой заболеваемостью раком про статы, смертность от рака этого орга на не очень велика. Необходимо под черкнуть, что на высокие показатели заболеваемости раком простаты в США влияет обнаруженный в резуль тате скрининга латентный рак, кото-
41
рый в отсутствие скрининга не выяв ляется, не дает симптомов, не про грессирует и никогда не приводит к смерти. Скринингом рака простаты с помощью теста на простатспецифический антиген (ПСА) объясняется стремительный рост заболеваемости между серединой 80-х и началом 90-х годов XX в. с дальнейшим таким же стремительным падением. Эти тен денции заболеваемости раком проста ты, характерные для США и других стран с высокой заболеваемостью, не сопровождались аналогичным по вы раженности ростом смертности. В этих регионах наблюдались лишь не большой рост или стабилизация пока зателей смертности.
Низкие показатели заболеваемости и смертности от рака простаты реги стрируют в странах Южной и Восточ ной Европы. Заболеваемость раком простаты очень низка в Юго-Восточ ной Азии.
У переселенцев из стран с низкой заболеваемостью, например японцев и китайцев, проживающих в США, за болеваемость значительно выше, чем в Японии и Китае. Однако она ниже, чем у "белых" и чернокожих амери канцев, что говорит о роли как факто ров окружающей среды, так и наслед ственности в этиологии рака этого ор гана.
Пятилетняя выживаемость от рака простаты в значительной степени за висит от наличия в стране скрининга и равна практически 100 % для ма леньких, латентных опухолей, выяв ленных при скрининге.
Рак шейки матки. В развивающих ся странах доля рака шейки матки среди всех злокачественных опухолей у женщин составляет 15 %, а в разви тых странах — 4 %. Самая высокая за болеваемость раком шейки матки ре гистрируется в Африке среди корен ного населения, Латинской Америке, Южной и Юго-Восточной Азии. В развитых странах заболеваемость низ кая. Смертность от рака шейки матки колеблется между самым высоким по казателем — более 10 на 100 тыс. на
селения в Мексике, Венесуэле, Тринидад и Тобаго и низким — 2 и меньше на 100 тыс. населения в Гре ции, Финляндии и Азербайджане.
Заболеваемость и смертность от рака шейки матки значительно снизи лись за последние десятилетия и про должают снижаться. Это особенно вы ражено в западных странах, где про водится массовый скрининг, с помо щью которого диагностируются ран ние формы рака и предрак шейки матки, что приводит к снижению не только смертности, но и заболеваемо сти раком этого органа.
Пятилетняя выживаемость при ра ке шейки матки довольно высока. Лучшие ее показатели (69 и 54 %) за регистрированы в США и Европе. В развивающихся странах этот показа тель равен 48 %.
Рак полости рта и глотки. Раком полости рта и глотки в два раза чаще болеют мужчины, чем женщины. Зло качественные опухоли полости рта и глотки объединены в одну группу вви ду схожести этиологии. Для всех них, кроме рака носоглотки, основными этиологическими факторами являют ся табак, включая оральные формы его потребления, жевание табака, и алкоголь. При раке же носоглотки до минирующим этиологическим факто ром является вирус Эпштейна—Барр. Последняя форма рака чаще встреча ется в Юго-Восточной Азии и крайне редко в Европе, Северной Америке и других развитых регионах мира.
Смертность от рака полости рта и глотки, так же как и рака легкого, наиболее высока в Восточной Европе, в частности в Венгрии, Словакии, Хорватии, Молдове. Самая низкая смертность зарегистрирована в Чили, Венесуэле, Швеции.
Пятилетняя выживаемость при этом заболевании в развитых странах равна 55 %. В регионах, где в структу ре рака полости рта и глотки домини рует рак носоглотки, показатели вы живаемости ниже.
Рак пищевода. Для рака пищевода характерна выраженная географиче-
42
екая вариабельность в заболеваемости
исмертности со 100 и более кратной разницей между высокой и низкой за болеваемостью. Самые высокие пока затели (> 150 на 100 тыс.) отмечены в Иране и странах так называемого Каспийского пояса, а именно: в неко торых районах Туркменистана и Ка захстана, прилегающих к Каспийско му морю, а также в Каракалпакии. Другими очагами очень высокой забо леваемости являются отдельные ре гионы Китая. Перечисленные выше регионы не имеют современных кан цер-регистров, не представлены в сборнике "Рак на пяти континентах",
иинформация о заболеваемости ра ком пищевода была получена в ре зультате специальных исследований. Среди представленных в табл. 1.2 стран высокая заболеваемость отмече на в Зимбабве, Бразилии и Китае. Этиология рака пищевода в этих эн демичных регионах остается неясной.
Вразвитых странах относительно высокая заболеваемость раком пище вода (> 10—15 на 100 тыс. населения) регистрируется среди чернокожего на селения в США, а также в Кальвадосе
инекоторых других регионах Фран ции. Основной причиной возникнове ния рака пищевода в развитых странах является курение и потребление алко голя.
Пятилетняя выживаемость раком пищевода колеблется от 10 % в США до 5 % в Европе.
Рак мочевого пузыря. Очень высо кие показатели заболеваемости раком мочевого пузыря отмечены в ряде Итальянских регистров, в некоторых регионах Испании. Заболеваемость также высока в большинстве стран Европы, в США и Израиле. Мужчины болеют раком мочевого пузыря чаще, чем женщины. Соотношение заболе ваемости среди мужчин к заболевае мости среди женщин в разных странах варьирует в пределах 5—35. Основной причиной рака мочевого пузыря явля ются курение, а также некоторые про фессиональные факторы. Однако в Северной Африке основной причиной
высокой заболеваемости раком моче вого пузыря является паразит Schistosoma mansoni и viverini.
Заболеваемость раком мочевого пузыря, как и других, вызванных ку рением форм рака, растет в большин стве стран мира. Пятилетняя выжи ваемость больных раком мочевого пу зыря очень высока в США (80 %) и значительно ниже в развивающихся странах. Такой высокий процент вы живаемости в США можно частично объяснить тем, что статистика заболе ваемости раком мочевого пузыря включает рак in situ (интраэпителиальный, или неинвазивный, рак).
Рак поджелудочной железы. Забо леваемость раком поджелудочной же лезы высокая в развитых странах и низкая в большинстве стран Африки и Азии, кроме Японии. Заболевае мость среди мужчин и женщин при мерно одинакова. Заболеваемость рас тет в странах с низкой заболеваемо стью и стабилизируется или снижается в странах с высокой заболеваемостью.
Прогноз при раке поджелудочной железы крайне неблагоприятный. Пя тилетняя выживаемость в развитых странах не превышает 5 %.
Рак яичника. Заболеваемость раком яичника высока в развитых странах. Однако разница в показателях заболе ваемости не очень выражена. Самые высокие показатели регистрируются в среде "белого" населения в Зимбабве, на Гавайях среди "белого" населения, в Дании, в Варшаве.
Заболеваемость и смертность в большинстве стран с высокой заболе ваемостью и смертностью стабильна или снижается, а в странах с низкой заболеваемостью отмечается рост этих показателей.
Рак тела матки. Географические особенности распространения рака тела матки такие же, как и при раке яичника. Высокая заболеваемость в США, Канаде и в большинстве евро пейских стран и низкая в развиваю щихся странах и Японии.
Заболеваемость и смертность от рака тела матки снижается в боль-
43
шинстве развитых стран, однако в развивающихся странах отмечается рост этого заболевания. Пятилетняя выживаемость в США равна 84 %, а в Европе — 12%.
Рак гортани. Рак гортани крайне редок среди женщин. Высокие пока затели заболеваемости (>10 на 100 тыс. населения) отмечают в Юго-Вос точной Европе, среди чернокожих в США, низкие — в Азии и Африке.
Смертность от рака гортани высока в Венгрии, Польше, Франции, Испа нии, и в большинстве стран мира на блюдается тенденция к росту данной патологии. Но в развитых странах, и прежде всего в США и Великобрита нии, смертность начала снижаться. Основными этиологическими факто рами рака гортани являются табак и алкоголь.
Меланома. Наиболее высокие по казатели заболеваемости и смертности от меланомы отмечены среди "белого" населения, проживающего в странах с жарким климатом: в Австралии, Но вой Зеландии, южных штатах США. Частота заболеваемости меланомой примерно одинакова среди мужчин и женщин. Отмечается выраженный рост частоты меланомы практически во всех странах мира. Пятилетняя вы живаемость очень высока в Австра лии, Новой Зеландии (85 %) и США (88 %). В Европе этот показатель ни же (70—75 %). В развивающихся же странах он равен примерно 50 %.
1.3. Основные факторы риска злокачественных опухолей
ипрофилактика
1.3.1.Курение и другие формы потребления табака
Влияние курения на риск возник новения злокачественных опухолей изучено досконально. На основании обобщения результатов эпидемиоло гических и экспериментальных иссле дований, а также изучения химиче ского состава табачного дыма рабочая группа МАИР, созванная в 1985 г.,
Т а б л и ц а 1.3. Факторы, канцерогенность
которых для человека доказана (группа 1)
Факторы |
Злокачественная |
|
опухоль |
||
|
||
Алкогольные на |
Рак полости рта, глот |
|
питки |
ки, гортани, пищевода, |
|
|
желудка, поджелудоч |
|
|
ной железы, печени |
|
Курение |
Рак полости рта, губы, |
|
|
глотки, гортани, пище |
|
|
вода, легкого, поджелу |
|
|
дочной железы, моче |
|
|
вого пузыря |
|
Табак некури |
Рак полости рта, носа, |
|
тельный |
носовых пазух |
|
Смесь бетеля с |
|
|
табаком |
|
|
Продукты сгора |
Рак кожи, мошонки, |
|
ния угля |
легкого, мочевого пу |
|
Каменноуголь |
зыря |
|
ная смола |
|
|
Деготь |
|
|
Сажа |
|
|
Минеральные |
Рак кожи, мошонки, |
|
масла |
легкого |
|
Сланцевые масла |
|
|
Соленая рыба |
Рак носоглотки, пище |
|
(китайского при |
вода, желудка |
|
готовления) |
|
|
Древесная пыль |
Рак полости и пазух |
|
Препараты, со |
||
носа |
||
держащие фен |
||
|
||
ацетин |
|
пришла к заключению, что курение табака является канцерогенным фак тором для человека и приводит к развитию рака губы, языка и других отделов полости рта, глотки, пищево да, поджелудочной железы, гортани, трахеи, бронхов, мочевого пузыря и почки (табл. 1.3). Табачный дым при нято делить на основную и побочную струю. Первая образуется при затяжке и вдыхается непосредственно куря щим, побочная же струя возникает при тлении сигареты между затяжка ми и попадает в окружающую среду.
Табак содержит никотин, который признан международными медицин скими организациями веществом, вы зывающим наркотическую зависи-
44
мость. Зависимость от табака внесена в международную классификацию бо лезней. Никотин соответствует ключе вым критериям наркотической зави симости, включая навязчивую, непре одолимую тягу к потреблению, не смотря на желание и повторяющиеся попытки отказаться от курения, пси хоактивные эффекты, развивающиеся при действии вещества на мозг, и осо бенности поведения, вызванные воз действием психоактивного вещества,
втом числе синдром абстиненции.
Всостав табачного дыма, кроме никотина, входит несколько десятков токсичных и канцерогенных веществ,
вих числе полициклические аромати ческие углеводороды (ПАУ), напри мер бенз(а)пирен, ароматические амины (нафтиламин, аминобифенил), летучие нитрозосоединения, табакспецифические нитрозоамины (ТСНА), винилхлорид, бензол, альдегиды (фор мальдегид), фенолы, хром, кадмий, по- лоний-210, свободные радикалы и т. д. Некоторые из этих веществ содержатся
втабачном листе, другие же образуют ся при его обработке и горении.
Необходимо подчеркнуть, что тем пература горения табака в сигаретах очень высока при затяжке и значи тельно ниже между затяжками, что определяет различную концентрацию химических веществ в основной и по бочной струе табачного дыма. В по бочной струе больше никотина, бен зола, ПАУ, чем в основной. Однако побочная струя быстро растворяется в воздухе, в результате в ней снижается концентрация химических, в том чис ле и канцерогенных веществ и соот ветственно канцерогенный потенциал.
Большинство канцерогенных и му тагенных веществ содержится в твер дой фазе табачного дыма, которая ос тается на так называемом кембридж ском фильтре при прокуривании сига рет на курительной машине. Смолой принято называть твердую фракцию табачного дыма, задержанную кем бриджским фильтром, без воды и ни котина. В зависимости от типа сига рет, фильтра, которым они снабжены,
сорта табака и его обработки, качества и степени перфорации сигаретной бу маги содержание смолы и никотина в табачном дыме может быть самым различным. За последние 20—25 лет произошло значительное снижение концентрации смолы и никотина в та бачном дыме сигарет, производимых в развитых странах, в том числе и Рос сии. В большинстве стран введены нормативы на содержание смолы и никотина. Для смолы эти нормативы варьируют в пределах 10—15 мг, а для никотина — 1—1,3 мг на 1 сигарету.
Канцерогенность табачного дыма показана в экспериментах на лабора торных животных. Контакт с табач ным дымом вызывает злокачествен ные опухоли гортани и легких. Одна ко трудность проведения подобных экспериментов с вдыханием табачного дыма очевидна ввиду невозможности имитации на животных процесса ку рения. Кроме того, как известно, про должительность жизни лабораторных животных, таких как мыши и крысы, очень коротка, что мешает постановке долгосрочных экспериментов, имити рующих длительный (20 лет и более) процесс канцерогенеза у человека.
Этиологическая связь между куре нием и злокачественными опухолями доказана во многих эпидемиологиче ских исследованиях, как когортных, так и методом случай—контроль. По казатель ОР, связанный с курением, разный при опухолях различных лока лизаций и зависит от возраста начала курения, длительности и количества сигарет, выкуриваемых в день.
Риск возникновения рака полости рта и глотки у курящих повышен в 2— 3 раза по сравнению с некурящими, а у выкуривающих более одной пачки сигарет в день относительный риск достигает 10.
Риск возникновения рака гортани
илегкого у курильщиков очень высок.
Вбольшинстве эпидемиологических когортных исследований отмечена дозовая зависимость между возрастом начала курения, длительностью, коли чеством сигарет, выкуриваемых в
45
день, и показателем ОР. Например, по данным когортного исследования английских врачей, ОР рака легкого равен 7,9 у курящих 1—14 сигарет, 12,7 — у выкуривающих 15—24 сига реты и 25 — у тех, кто курит более 25 сигарет в день. Результаты когортного исследования американского проти воракового общества и когортных ис следований, проведенных в других странах, доказывают важную роль возраста начала курения. Наибольший ОР рака легкого отмечен у мужчин, которые начали курить до 15 лет (15,0). У мужчин, начавших курить в возрасте 15—19, 20—24 и более 25 лет, ОР был равен 12,8, 9,7 и 3,2 соответ ственно. Следует отметить, что этио логическая связь между курением и раком легкого более выражена для плоскоклеточного и мелкоклеточного рака, чем для аденокарциномы.
Риск возникновения рака пищево да в 5 раз выше у курящих по сравне нию с некурящими. Риск возникнове ния рака желудка у курящих несколь ко ниже и равен 1,3—1,5. Курение яв ляется одной из причин рака подже лудочной железы. ОР возникновения рака поджелудочной железы у куря щих повышен в 2—3 раза. Курение, по-видимому, не влияет на риск рака ободочной и прямой кишки, однако в ряде эпидемиологических исследова ний выявлена ассоциация между ку рением и аденоматозными полипами толстой кишки. Связь между курени ем и возникновением рака ануса, опу холью, имеющей плоскоклеточное или переходноклеточное строение, была показана неоднократно.
В некоторых эпидемиологических исследованиях выявлен повышенный риск печеночно-клеточного рака, свя занный с курением. Курение в сочета нии с потреблением алкоголя повы шает риск гепатоцеллюлярного рака печени даже при отсутствии инфек ции вирусом гепатита В. Связи между курением и холангиоцеллюлярным раком, а также злокачественными опухолями желчного пузыря и желч ных протоков не обнаружено.
Курение является причиной разви тия рака мочевого пузыря и почки. Риск рака мочевого пузыря среди ку рящих повышен в 5—6 раз. Связь ме жду курением и риском рака почки показана как для плоскоклеточного и переходноклеточного рака, так и для аденокарциномы.
Выявлена зависимость рака шейки матки и интраэпителиальной неоплазии с курением. Учитывая тот факт, что инфицированность вирусом па пилломы человека (ВПЧ) является до казанной причиной рака шейки мат ки, курение скорее всего играет роль промотора процесса канцерогенеза в шейке матки, инфицированного ВПЧ.
Рак тела матки является единст венной формой рака, риск которого у курящих женщин снижен. Это наблю дение подтверждено в нескольких ис следованиях методом случай—кон троль. Показатель относительного риска рака эндометрия у курящих женщин равен 0,4—0,8. Защитный эф фект курения против рака этой лока лизации можно скорее всего объяс нить гормональным механизмом — снижением (ингибированием) продук ции эстрогенов. Кроме того, у куря щих женщин менопауза наступает на 2—3 года раньше, чем у некурящих.
Курение, по-видимому, не влияет на развитие рака яичников. В то же время существует связь между курени ем и риском рака вульвы. Курение как причина рака молочной железы изу чено во многих эпидемиологических исследованиях, результаты которых указывают, что курение скорее всего не способствует развитию рака молоч ной железы. Рак простаты относится к формам рака, на риск которого куре ние, по всей видимости, не влияет. В ряде эпидемиологических исследова ний выявлена связь курения с ОР лейкозов, в частности ОР острого миелобластного лейкоза у курящих равен 1,5.
Атрибутивный риск (АР), т. е. про цент всех случаев рака, этиологически связанный с курением, различен для различных форм злокачественных
46
опухолей. Например, по самым кон |
Как |
известно, |
возрастная |
кривая |
||||||||||||
сервативным оценкам, непосредствен |
смертности |
от |
ишемической |
болезни |
||||||||||||
ной причиной 85—90 % рака легкого у |
сердца достигает своего пика значи |
|||||||||||||||
мужчин является курение сигарет. По |
тельно раньше, чем для рака легкого. |
|||||||||||||||
данным Р. Долла и Р. Пето (1988), ос |
Курящие мужчины в этой популяции |
|||||||||||||||
нованным |
на статистике |
злокачест |
умиряют в относительно раннем воз |
|||||||||||||
венных опухолей и на эпидемиологи |
расте от ишемической болезни сердца |
|||||||||||||||
ческих исследованиях, |
проведенных в |
и |
других |
конкурирующих |
причин |
|||||||||||
США, курение сигарет является при |
смерти, не достигая возраста, когда |
|||||||||||||||
чиной 35 % всех злокачественных |
начинает расти смертность от рака |
|||||||||||||||
опухолей. Аналогичные |
оценки |
роли |
легкого, т. е. они попросту не дожива |
|||||||||||||
курения в этиологии злокачественных |
ют до своего рака легкого. Этим, кста |
|||||||||||||||
опухолей сделаны и для других разви |
ти, можно частично объяснить и сни |
|||||||||||||||
тых стран с высоким процентом куря |
жение смертности от рака легкого |
|||||||||||||||
щих и длительной историей курения. |
среди российских мужчин. |
|
||||||||||||||
В |
исследовании, проведенном на |
Несмотря |
на |
распространенное |
||||||||||||
ми в четырех городах России. Барнау |
мнение, что курение сигар не являет |
|||||||||||||||
ле, Владивостоке, Томске и Тюмени, — |
ся канцерогенным фактором, получе |
|||||||||||||||
были |
опрошены |
ближайшие |
родст |
ны убедительные эпидемиологические |
||||||||||||
венники 7800 умерших мужчин и 5300 |
данные, что курение сигар повышает |
|||||||||||||||
женщин. Необходимо отметить очень |
риск рака полости рта, глотки, горта |
|||||||||||||||
высокую частоту курения в этой попу |
ни, легкого, пищевода и поджелудоч |
|||||||||||||||
ляции. Среди мужчин, умерших от |
ной |
железы. |
Причем выраженность |
|||||||||||||
причин, связанных с курением, она |
канцерогенного эффекта сигар на по |
|||||||||||||||
достигала 63 %. Особенно высока бы |
лость рта, глотку и гортань аналогич |
|||||||||||||||
ла частота курения в возрастных груп |
на эффекту сигарет. Риск рака легкого |
|||||||||||||||
пах 25—34, 35—44, 45—54, где курили |
у курящих сигары несколько ниже, но |
|||||||||||||||
соответственно 76, 82 и 77 % мужчин. |
может достигать |
высоких показателей |
||||||||||||||
В этом исследовании было показано, |
у тех, кто глубоко затягивается. Отно |
|||||||||||||||
что ОР смерти от рака полости рта, |
сительный риск злокачественных опу |
|||||||||||||||
глотки, пищевода и гортани равен 4,4, |
холей у курящих зависит от длитель |
|||||||||||||||
а АР среди курящих — 78 %. ОР смер |
ности |
курения, |
количества |
выкури |
||||||||||||
ти от рака легкого, связанный с куре |
ваемых сигар в день, а также от того, |
|||||||||||||||
нием, был 4,5, а АР среди курящих |
совмещается ли курение сигар с куре |
|||||||||||||||
мужчин — 78 %. Очевидно, что пока |
нием сигарет или трубки. Сигарный |
|||||||||||||||
затели ОР и АР смерти от рака легко |
дым содержит практически все те же |
|||||||||||||||
го, связанные с курением, в нашем |
токсичные |
и |
канцерогенные |
вещест |
||||||||||||
исследовании ниже, чем в описанных |
ва, что и табачный дым сигарет, одна |
|||||||||||||||
выше эпидемиологических исследова |
ко в нем больше никотина и ТСНА. |
|||||||||||||||
ниях, проведенных в США и Англии. |
Кроме того, рН сигарного дыма вы |
|||||||||||||||
В то же время эти показатели значи |
ше, чем у сигаретного дыма, что явля |
|||||||||||||||
тельно выше для ишемической болез |
ется препятствием, хоть и относитель |
|||||||||||||||
ни сердца (ОР — 2,0; АР - 49 %), ин |
ным, к его вдыханию. Никотин и дру |
|||||||||||||||
сульта (ОР — 2,1; АР — 53 %), других |
гие вещества всасываются через сли |
|||||||||||||||
сердечно-сосудистых |
заболеваний |
зистую оболочку полости рта, а если |
||||||||||||||
(ОР - 1,5; АР — 34 %), а также ин |
курильщик все же затягивается, то и |
|||||||||||||||
фекционных |
заболеваний |
легких, в |
через слизистую оболочку бронхов. |
|||||||||||||
том числе и туберкулеза. Скорее всего |
На основании нескольких десятков |
|||||||||||||||
низкие показатели рисков, связанных |
||||||||||||||||
эпидемиологических |
исследований, |
|||||||||||||||
с курением, |
для рака легкого можно |
|||||||||||||||
проведенных за последние 10—15 лет, |
||||||||||||||||
объяснить |
очень |
высокой |
смертно |
|||||||||||||
можно сделать |
заключение, |
что пас |
||||||||||||||
стью в России от сердечно-сосудистых |
||||||||||||||||
сивное курение также является канце |
||||||||||||||||
заболеваний |
среди |
курящих мужчин. |
||||||||||||||
рогенным для человека. ОР рака лег- |
||||||||||||||||
|
|
|
|
|
|
|
|
|||||||||
47
кого у некурящих женщин, мужья ко торых курят, равен по данным различ ных исследований 1,3—1,7. В исследо вании методом случай—контроль, проведенном нами в Москве среди некурящих женщин, мужья которых курят, ОР рака легкого достигал 1,7 и зависел от длительности курения му жа и количества выкуриваемых им в день сигарет. Агентство по защите ок ружающей среды США пришло к за ключению, что пассивное курение яв ляется причиной смерти от рака лег кого 3000 американцев в год и что пассивное курение повышает риск возникновения рака легкого на 30 % (т. е. ОР равен 1,3).
Таким образом, курение является важнейшей причиной развития злока чественных опухолей. Снижение час тоты курения среди населения неко торых развитых стран, например США и Великобритании, уже привело к уменьшению заболеваемости и смертности от рака легкого и других форм рака, этиологически связанных с курением. Нужно также отметить, что снижение частоты курения в этих странах значительно раньше сказалось на смертности от сердечно-сосуди стых заболеваний.
Кроме курения, известны и другие формы потребления табака. В Индии табак и различные его смеси (напри мер, смесь табака с известью или по рошком измельченных ракушек, за вернутых в лист бетеля) закладывают за щеку и под язык или жуют. В стра нах Центральной Азии распространен нас (нос), который представляет из себя смесь табака с известью и золой, его тоже закладывают под язык или за щеку. В Швеции распространен та бачный продукт снус, который также предназначен для перорального по требления. Кроме того, существуют и нюхательные табаки.
В отличие от табачного дыма пере численные выше типы табачных изде лий не содержат канцерогенных ве ществ, которые образуются в резуль тате горения табака при высоких тем пературах. Однако в их состав входят
табакоспецифические нитрозоамины (ТСНА), такие как N-нитрозонорни- котин (NNN), 4-метилнитрозоамино- 1-(3-пиридил)-1-бутанон (NNK), канцерогенность которых доказана. Эпи демиологические исследования пока зали, что потребление орально табач ных изделий повышает риск развития рака полости рта и глотки. Кроме то го, выявлена связь между потреблени ем оральных форм табака и наличием лейкоплакий, патологических образо ваний слизистой оболочки полости рта, которые обычно предшествуют развитию рака.
Рабочая группа МАИР, созванная в 1984 г., на основании анализа экспе риментальных и эпидемиологических данных пришла к заключению, что оральные формы табачных изделий являются канцерогенными для чело века и их потребление приводит к развитию рака полости рта и глотки.
1.3.2. Питание
Питание играет важную роль в этиологии злокачественных опухо лей, по крайней мере одна треть всех злокачественных опухолей связана с питанием. Известно, что заболевае мость и смертность от злокачествен ных опухолей значительно варьирует в различных географических регионах, например заболеваемость раком же лудка очень высока в Японии, Корее, Китае и низка в Северной Америке. В то же время заболеваемость злокаче ственными опухолями толстой киш ки, молочной железы, простаты низка в странах Юго-Восточной Азии и вы сока в Северной Америке и Западной Европе.
Исследования мигрантов из стран Юго-Восточной Азии в США (Гавайи
иКалифорнию) показали, что у япон цев и китайцев, проживающих в США, уже в первом поколении сни зилась заболеваемость раком желудка
ивыросла заболеваемость раком тол стой кишки. Более того, самая высо кая заболеваемость раком толстой кишки в мире зарегистрирована у
48
японцев, проживающих на Гавайях, в то время как у японцев, проживаю щих в Японии, заболеваемость раком толстой кишки хоть и растет, но оста ется значительно ниже показателей в Северной Америке и Европе. Заболе ваемость раком молочной железы и простаты у мигрантов из Юго-Восточ ной Азии, проживающих в США, так же велика, но ниже, чем у "белых" и "черных" американцев.
Исследования мигрантов позволи ли предположить, что географическая вариабельность в заболеваемости зло качественными опухолями обусловле на некими факторами окружающей среды и образа жизни, а не популяционными генетическими особенностя ми, в связи с чем и была сформулиро вана гипотеза о роли питания в этио логии злокачественных опухолей.
Наблюдения за некоторыми рели гиозными группами, например адвен тистами Седьмого дня, придерживаю щимися особой диеты, не включаю щей мясных продуктов, показало, что у них заболеваемость раком толстой кишки, молочной железы, тела матки и простаты значительно ниже, чем у остального населения, проживающе го рядом с ними.
Связь между особенностями пита ния и заболеваемостью злокачествен ными опухолями была впервые пока зана в корреляционных исследовани ях. Было выявлено, что потребление жиров, а особенно животных, мяса и молока, количество потребляемых ка лорий коррелирует с заболеваемостью раком толстой кишки, молочной же лезы, матки и простаты. Эксперимен тальные исследования показали, что ограничение потребления калорий, а также насыщенных жиров животного происхождения ингибирует процесс канцерогенеза, индуцированный хи мическими канцерогенными вещест вами. В некоторых эксперименталь ных исследованиях снижение потреб ления жиров, без соответствующего снижения потребляемых калорий, приводило к уменьшению количества индуцированных опухолей и удлине
нию латентного периода их развития, т. е. ингибированию процесса канце рогенеза. Ингибирование процесса канцерогенеза, индуцированного хи мическими канцерогенными вещест вами в результате ограничения по требления энергии и животных жи ров, было отмечено для злокачествен ных опухолей молочной железы и тол стой кишки, а также легкого, кожи и некоторых неэпителиальных опухо лей. Необходимо отметить, что как в эксперименте, так и в эпидемиологи ческих исследованиях очень трудно полностью разграничить влияние по требления калорий от потребления жиров, так как животный жир являет ся наиболее энергоемким компонен том питания и основным источником калорий.
Механизм ингибирования опухоле вого роста, связанный с ограничением потребляемых калорий, можно объяс нить снижением пролиферации кле ток и стимулированием апоптоза, уси лением репарации ДНК, снижением образования свободных радикалов и соответственно повреждения ими кле ток, изменением гормонального про филя, в частности снижением уровня как общего, так и свободного эстрадиола и тестостерона.
Маркерами потребляемой энергии в детстве и во взрослом возрасте явля ются рост (темп роста), масса тела, а также уровень физической активно сти. У женщин очень важным марке ром потребляемых калорий в детском возрасте является и возраст начала менструации. Эпидемиологические исследования показали, что все пере численные характеристики влияют на риск развития рака.
Доказано, что ожирение является доминирующим фактором риска для рака эндометрия, он увеличивается примерно в три раза при повышении весоростового индекса с 20 до 35. Кроме того, избыточная масса тела повышает риск рака желчного пузыря, скорее всего рака молочной железы и почки, а также толстой кишки. Для рака молочной железы показано уд-
4-7908 Д. Г. Заридзе |
49 |
воение ОР при повышении весоростового индекса с 17 до 37.
Что же касается физической актив ности, то достоверно доказано, что повышенная физическая активность, как профессиональная, так и связан ная со спортивными занятиями, сни жает риск развития рака ободочной кишки на 60 %. В одном исследова нии когорты американских женщин было показано, что у тех из них, кото рые в молодости активно занимались спортом, снижен риск практически всех гормонозависимых форм рака — молочной железы, эндометрия и яич ника.
Быстрый рост, который, как и мас са тела, отражает высокий уровень по требления калорий (только в детстве и подростковом возрасте) и раннее на чало менструаций, являются доказан ными факторами риска рака молоч ной железы, возможно, и рака тол стой кишки. Соответственно неболь шой рост и позднее начало менструа ции снижает ОР развития рака молоч ной железы и других органов женской репродуктивной системы.
Эпидемиологические исследова ния, проведенные в США, показали, что наиболее низкие показатели смертности от всех форм злокачест венных опухолей были у мужчин, мас са тела которых была между 10 % ни же и 20 % выше среднего показателя американцев. У женщин наиболее благоприятная масса тела в пределах 20 % ниже и 10 % выше среднего веса американок.
Механизм канцерогенного эффек та жиров связывают с их влиянием на процессы синтеза и метаболизма сте роидных половых гормонов, таких как эстрадиол и тестостерон. Жирные ки слоты, особенно насыщенные, ингибируют связывание эстрадиола "стеро идные половые гормоны связываю щим глобулином" (СПГСГ), что явля ется причиной высокой концентрации циркулирующего в крови свободного эстрадиола. Было показано, что уменьшение потребления жиров при водит к снижению уровней эстрона и
эстрадиола у женщин детородного возраста. У женщин в менопаузе уменьшение потребления жиров с 40 до 20 % потребляемых калорий приве ло к выраженному (на 17 %) сниже нию концентрации в плазме крови общего эстрадиола. На основании анализа результатов исследований ме тодом случай—контроль американ ские ученые пришли к заключению, что снижение на 17 % концентрации общего эстрадиола в крови приводит к 4—5-кратному снижению риска рака молочной железы. Потребление жи ров влияет также на концентрацию мужского полового гормона тестосте рона. Доказано, что концентрация в крови тестостерона достоверно корре лирует с потреблением жиров. Напри мер, концентрация в крови тестосте рона значительно выше у американ ских африканцев, чем африканцев, проживающих в Африке: у последних значительно ниже потребление жиров. В то же время заболеваемость раком простаты значительно выше у амери канских африканцев. Исследование гормонального профиля больных ра ком простаты и контрольной группы в странах с низкой и высокой заболе ваемостью раком простаты (Японии и Нидерландах) показало, что как по требление жиров, так и концентрация тестостерона в крови значительно вы ше у голландцев.
Механизм действия жиров на про цесс канцерогенеза в толстой кишке связан с их влиянием на метаболизм кишечной флоры и концентрацию вторичных жирных кислот, которые являются промоторами канцерогене за у лабораторных животных. Кроме того, жиры стимулируют образование в толстой кишке фекапентанов — ве ществ, обладающих мутагенным дей ствием, и скорее всего очень важны для процесса канцерогенеза в этом органе. В результате переработки жи ров в кишечнике образуются фекаль ные стеролы, некоторые из которых играют ключевую роль в пролифера ции толстокишечного эпителия. По казано, что у людей с высоким по-
50
треблением жиров в кале отмечается высокая концентрация вторичных жирных кислот, фекапентанов, более выражен метаболизм кишечной фло ры, а также и процесс превращения липидов в мутагенные фекальные стеролы.
В эпидемиологических исследова ниях, в которых изучается роль пита ния в этиологии того или иного забо левания, в том числе и рака, особен ности питания человека оцениваются с помощью специальных анкет, в ко торых сформулированы вопросы о частоте и количестве потребления ос новных продуктов питания — хлеба, молока, мяса, рыбы, овощей, фруктов и т. д. В дальнейшем полученные дан ные с помощью специальных таблиц переводят в нутриенты: жиры (насы щенные, мононенасыщенные, поли ненасыщенные), белки (животного и растительного происхождения), угле воды, микроэлементы, витамины и т. д. Рассчитывается количество по требляемых калорий. Результаты та ких исследований обычно представле ны как в единицах продуктов, так и в единицах нутриентов и микроэлемен тов. Характер питания также можно оценить с помощью прямого анализа в крови или других биологических ма териалах, маркеров потребления тех или иных нутриентов или микроэле ментов, например жирных кислот, большинства витаминов и металлов, в частности селена. Такие исследования принято называть метаболическими эпидемиологическими исследованиями.
Связь между потреблением живот ных жиров и риском рака толстой кишки, молочной железы, простаты была показана во многих аналитиче ских эпидемиологических исследова ниях. В большинстве работ, опубли кованных до середины 80-х годов XX в., было выявлено, что риск рака тол стой кишки, молочной железы, про статы повышен у людей с высоким потреблением животных жиров и мя са. Однако аналитические эпидемио логические исследования последую щих лет, в которых использовались
более точные методы оценки потреб ления жиров и других нутриентов и усовершенствованные методы стати стического анализа, внесли сомнения
всуществование причинной связи ме жду потреблением жиров вообще и насыщенных жиров в частности и риском рака этих органов. Когортное исследование американских медсе стер, которое включало 90 тыс. участ ников, не выявило повышения риска рака молочной железы в группе жен щин с высоким потреблением жира вообще и в частности насыщенных жирных кислот, линолевой кислоты и холестерина. Однако мета-анализ 12 исследований методом случай—кон троль показал статистически досто верное повышение риска рака молоч ной железы, связанное с высокими показателями потребления жира вооб ще и насыщенных жиров в частности.
Вто же время мета-анализ 13 других эпидемиологических исследований не выявил связи между потреблением жиров и раком молочной железы. Та ким образом, роль потребления жиров
вэтиологии рака молочной железы остается недоказанной. Высказывает ся предположение, что в тех исследо ваниях, где такая связь выявлена, ско рее всего не удалось отделить эффект потребления энергии от эффекта по требления жиров. Как уже говорилось выше, жир является наиболее энерго емким нутриентом и большая часть потребляемых человеком калорий (бо лее 40 %), особенно в развитых стра нах, представлена жирами.
Что же касается эпидемиологиче ских исследований рака толстой киш ки, то в большинстве из них показана связь между потреблением жиров, особенно насыщенных, а также мяса с риском развития рака этого органа. Например, в уже упомянутом когортном исследовании американских мед сестер было выявлено статистически достоверное повышение риска рака ободочной кишки у женщин с высо ким потреблением животных жиров, говядины, свинины, баранины и кол басных изделий. В этом и другом аме-
4* |
51 |
риканских когортных исследованиях медработников было показано, что риск рака и аденоматозных полипов ободочной кишки зависит от соотно шения потребления мяса к потребле нию птицы и рыбы: чем выше потреб ление мяса по сравнению с потребле нием птицы и рыбы, тем выше риск рака этого органа.
Углеводы наряду с жирами являют ся важным источником калорий. В развивающихся странах углеводы со ставляют более 70 % потребляемой энергии. В развитых странах доля уг леводов в рационе питания снижается за счет роста потребления жиров. В продуктах питания углеводы представ лены в виде крахмала, Сахаров и дру гих полисахаридов, большая часть ко торых составляет так называемую клетчатку. Основным источником крахмала являются злаки (хлеб), кру пы, картофель, горох, бобы. Клетчат ка является неотъемлемым компонен том растительной пищи, овощей, фруктов и нерафинированных (неочи щенных) круп.
Гипотеза о защитной роли клетчат ки была сформулирована английским врачом Д. П. Беркиттом на основании наблюдений в Африке, где заболевае мость раком толстой кишки низка, а потребление продуктов питания с вы соким содержанием клетчатки высо ко. Предполагается, что у людей, по требляющих много клетчатки, увели чен объем каловых масс, что ведет к снижению в толстой кишке концен трации канцерогенных веществ.
Клинические метаболические ис следования показали, что добавление к ежедневному рациону 10—13 г цел люлозы или клетчатки зерновых (wheat bran) значительно снижает концентрацию в кале вторичных желчных кислот, их метаболическую и мутагенную активность.
Большинство аналитических эпи демиологических исследований под твердили гипотезу о протективном эффекте клетчатки. Мета-анализ 16 исследований методом случай—кон троль подтвердил обратную связь ме
жду потреблением клетчатки и пищи, богатой клетчаткой, и риском рака ободочной кишки. Проспективное ис следование американских медработ ников, в котором под наблюдением находились около 10 тыс. мужчин, выявило, что потребление клетчатки, источником которой являются фрук ты и овощи, а также зерновые и кру пы, снижает риск аденоматозных по липов и рака ободочной кишки. Од нако в когортном исследовании аме риканских медсестер, как и ряде дру гих эпидемиологических исследова ний, было показано, что протективным эффектом против рака толстой кишки обладает лишь клетчатка ово щей и фруктов. Этот защитный эф фект может быть результатом дейст вия витаминов, индолов, протеаз и других компонентов фруктов и ово щей.
Защитное влияние потребления овощей и фруктов против развития злокачественных опухолей у человека доказано для рака полости рта и глот ки, пищевода, желудка, ободочной и прямой кишки, меньше эпидемиоло гических научных доказательств для злокачественных опухолей гортани, легкого, поджелудочной железы, мо лочной железы и мочевого пузыря. Однако потребление овощей и фрук тов скорее всего снижает риск разви тия этих опухолей. В некоторых эпи демиологических исследованиях сни жение ОР в связи с потреблением овощей и фруктов было отмечено и для опухолей шейки матки, эндомет рия, почки и простаты.
Кроме того, доказано, что потреб ление овощей и фруктов снижает риск всех форм злокачественных опу холей в целом. В одном когортном ис следовании выявлено, что у мужчин, которые ели много зеленых и желтых овощей, риск смерти от всех форм ра ка был равен 0,3.
Выраженным защитным эффек том обладают лук и чеснок. В иссле довании, проведенном нами в Моск ве, было показано, что потребление чеснока значительно снижает риск ра-
52
ка желудка. Антиканцерогенный эф фект чеснока можно объяснить его бактерицидными свойствами, в част ности против Helicobacter pylori, инфицированность которым является известным фактором риска рака же лудка. Овощи и фрукты содержат ак тивные вещества, которые в экспери менте на лабораторных животных ингибируют развитие опухолей. К ним в первую очередь относятся витамины С, Е, бета-каротин, селен, обладаю щие антиоксидантными свойствами, витамин А, фолиевая кислота, а также фитоэстрогены (изофлавинолы), флавоноиды, такие как кверцетин, индо лы и т. д.
Витамин А играет основную роль в дифференцировке клеток, что послу жило основанием для гипотезы о том, что витамин А может быть ингибито ром канцерогенеза. Эта гипотеза под тверждена в экспериментальных ис следованиях. Предшественниками ви тамина А являются каротиноиды, ко торые в эксперименте оказались ин гибиторами канцерогенеза, особенно на модели рака кожи. Аналитические эпидемиологические исследования подтвердили протективный эффект каротиноидов и в меньшей степени витамина А. Необходимо подчерк нуть, что источником витамина А яв ляются только продукты животного происхождения, в то время как каро тиноиды поступают в организм чело века исключительно с продуктами растительного происхождения.
В исследованиях методом случайконтроль и когортных исследованиях, когда оценивалось потребление каро тиноидов и бета-каротина с помощью анкеты, а также проводился анализ крови на содержание этих витаминов, выявлено, что высокий уровень по требления каротиноидов с пищей и высокая их концентрация в крови снижают риск рака легкого. Потреб ление витамина А и каротиноидов уменьшает риск рака гортани, пище вода, желудка, молочной железы, мо чевого пузыря, шейки матки.
Несмотря на достаточно убеди
тельные данные аналитических эпиде миологических исследований о протективном влиянии каротиноидов и бета-каротина, контролируемые ран домизированные исследования, в ко торых изучался эффект бета-каротина для профилактики рака, не дали ожи даемого результата. У больных раком кожи прием бета-каротина не привел к снижению частоты вторых опухолей кожи. Аналогично прием бета-кароти на, витамина С и Е не снизил частоту рецидивов аденоматозных полипов толстой кишки. Особенно следует подчеркнуть результаты крупных ран домизированных контролируемых ис следований, проведенных в США и Финляндии, по профилактике рака легкого среди курящих мужчин. В группе, получавшей бета-каротин, не было отмечено снижение риска рака легкого, а в одном из них этот риск был выше в опытной, чем в контроль ной группе.
Витамин С является антиоксидантом и, кроме того, ингибирует эндо генное образование в желудке нитрозаминов из поступающих с пищей аминов и нитритов. В ряде исследова ний методом случай—контроль отме чен протективный эффект потребле ния витамина С. Показано, что у лю дей, потребляющих с пищей много витамина С, снижен риск рака полос ти рта, гортани, пищевода, желудка и шейки матки. Как и в отношении других витаминов, остается не до кон ца ясно, обладают ли протективный эффектом витамин С или другие ком поненты фруктов и овощей, в состав которых входит витамин С.
Витамин Е также является мощ ным антиоксидантом. В эксперимен тальных исследованиях показано, что витамин Е ингибирует процесс канце рогенеза. Результаты эпидемиологиче ских исследований, в которых изуча лось влияние потребления витамина Е с пищей и его концентрации в крови, противоречивы. Однако следует отме тить, что в исследованиях, где изуча лась связь с концентрацией в крови витамина Е, была показана обратная
53
зависимость между уровнем витамина Е и риском возникновения злокачест венных опухолей и особенно тех, ко торые причинно не связаны с курени ем. В рандомизированном контроли руемом исследовании, проведенном в Финляндии, прием витамина Е не по влиял на риск рака легкого, однако в опытной группе был снижен риск ра ка простаты. В целом протективный эффект витамина Е скорее всего весь ма ограничен.
Эффект селена, ингибирующий канцерогенез, был показан множест вом экспериментальных исследова ний. Кроме того, эпидемиологически ми исследованиями отмечена обрат ная корреляция между уровнем по требления селена и заболеваемостью злокачественными опухолями. Корре ляция особенно выражена для рака толстой кишки и молочной железы. Результаты аналитических эпидемио логических исследований менее убе дительны. В некоторых проспектив ных исследованиях, в которых изуча лась концентрация селена в крови, выявлено снижение риска злокачест венных опухолей молочной железы и легкого с увеличением концентрации селена в крови. Что же касается дру гих форм рака, то эпидемиологиче ские исследования не могут ни под твердить, ни опровергнуть возмож ность защитного влияния селена на их развитие.
В соленых, копченых и консерви рованных продуктах могут содержать ся различные канцерогенные вещест ва. Есть основания предполагать, что нитрозамины, а также их предшест венники (нитраты, нитриты), находя щиеся в пище, повышают риск рака пищевода и желудка. Увеличивается риск рака желудка у людей, потреб ляющих много соли.
Несмотря на то что в настоящее время наших знаний недостаточно для того, чтобы точно указать на все ком поненты питания, способствующие развитию рака или, наоборот, сни жающие риск его развития, не вызы вает сомнения, что изменение пита
ния в сторону увеличения потребле ния овощей, зелени и фруктов, умень шения потребления жира, особенно насыщенного, и пищи, богатой жи ром, приведет к снижению заболевае мости злокачественными опухолями.
1.3.3.Эндогенные
иэкзогенные гормоны
Гормональный статус является фактором, определяющим риск мно гих злокачественных опухолей и пре жде всего рака тела матки, яичников, молочной железы, простаты и яичка. Скорее всего злокачественная опухоль развивается в результате повышенной (чрезмерной) гормональной стимуля ции органа, нормальный рост, разви тие и функция которого находятся под контролем того или иного стеро идного или полипептидного гормона. У женщин уровень эстрогенной сти муляции, или, если угодно, суммар ный уровень эстрогенной стимуляции в течение жизни, зависит от возраста менархе и менопаузы и от количества овуляций, последнее же в свою оче редь определяется количеством бере менностей. Беременность, как и, впрочем, оральные контрацептивы, содержащие прогестерон, приводят к супрессии овуляции и, соответственно снижению эстрогенной стимуляции гормонозависимых органов.
В мире отмечается значительное отличие в гормональном статусе раз ных групп населения, которая выра жается в разнице возраста менархе, менопаузы, роста и других конститу циональных особенностей организма. Это отличие обусловлено как наслед ственными, так и приобретенными особенностями организма. Послед ние в значительной степени зависят от особенностей образа жизни, осо бенно питания, и окружающей среды, а также климатических и экологиче ских факторов. С другой стороны, факторы репродуктивного анамнеза, такие как возраст первых родов, коли чество родов, применение пероральных контрацептивов и других гормо-
54
нальных препаратов, влияют на гор мональный статус и в первую очередь на суммарный уровень эстрогенной стимуляции в течение жизни.
Эндогенные гормоны. Синтез и метаболизм стероидных половых гор монов, как было сказано выше, в значительной степени определяются типом питания, в частности потреб лением жира. Жирные кислоты, осо бенно насыщенные, ингибируют свя зывание СПГСГ эстрадиола, что яв ляется причиной высокой концен трации в плазме свободного эстра диола. Показано, что у женщин дето родного возраста снижение потребле ния жира приводит к снижению кон центрации в крови эстрона и эстра диола. У женщин в менопаузе сниже ние в рационе жиров с 40 до 20 % потребляемых калорий привело к 17 % снижению в плазме концентра ции эстрадиола. Расчеты, основан ные на результатах эпидемиологиче ских исследований, показали, что снижение концентрации эстрадиола в плазме на 17 % должно привести к 4—5-кратному снижению риска рака молочной железы.
Изучение роли эндогенных стеро идных половых гормонов показало, что у женщин, больных раком молоч ной железы, концентрация в крови общего и свободного эстрадиола, т. е. не связанного с СПГСГ эстрадиола, выше, чем у здоровых женщин соот ветствующего возраста. Эта разница в концентрации эстрадиола наиболее выражена для женщин в менопаузе, у которых риск рака молочной железы статистически достоверно связан с концентрацией этих гормонов. Кроме того, в некоторых исследованиях вы явлена связь между концентрацией или уменьшением связывающей спо собности СПГСГ и риском рака мо лочной железы. При высокой концен трации и высокой способности СПГСГ связывать эстрадиол снижает ся концентрация свободной его фрак ции, что приводит к снижению риска рака молочной железы. Высокие уров ни эндогенных половых гормонов в
крови повышают риск рака тела мат ки и яичников.
Потребление жиров влияет и на концентрацию в крови мужского по лового гормона тестостерона. Кон центрация тестостерона в крови дос товерно коррелирует с потреблением жиров. Например, как концентрация тестостерона в крови, так и потребле ние жиров значительно выше у афроамериканцев, чем у африканцев, живу щих в Африке. У последних значи тельно ниже и заболеваемость раком простаты. Исследование, проведенное
врегионах с низкой и высокой забо леваемостью раком простаты, в Япо нии и Нидерландах, показало, что по требление жира и уровни тестостерона
вкрови достоверно выше у мужчин с высокой заболеваемостью раком про статы — у голландцев. Результаты эпидемиологических исследований, в которых изучалась связь между эндо генными мужскими половыми гормо нами и раком простаты, неубедитель ны. В исследованиях методом слу чай—контроль показано, что высокая концентрация в крови андрогенов, в частности тестостерона, повышает ОР рака простаты. Однако этот результат не подтвержден в когортных исследо ваниях, в которых, как и предполагает метод, кровь бралась у еще здоровых мужчин, у которых впоследствии (че рез несколько лет) развивался рак простаты. Мета-анализ когортных ис следований показал, что ОР рака про статы не связан с высокой концентра цией в крови эндогенных половых гормонов, за исключением андростандиол глюкоронида, концентрация ко торого у больных раком простаты бы ла на 5 % выше, чем у здоровых муж чин, членов той же когорты (ОР 1.05). Несоответствие между результатами исследований методом случай—кон троль и когортными исследованиями можно объяснить тем, что при первом методе кровь бралась у мужчин, у ко торых уже был рак простаты — бо лезнь, которая, по всей вероятности, повлияла на метаболизм гормонов, и высокая концентрация тестостерона у
55
них была не причиной, а следствием заболевания.
Репродуктивный анамнез. Репро дуктивный анамнез играет важную роль в этиологии опухолей женских половых органов. Ранний возраст на чала менструации (менархе) и поздняя менопауза повышают риск рака мо лочной железы, тела матки и яичника. В исследовании, проведенном нами в Москве, и ряде других исследований показано, что у женщин, возраст ме нархе которых был 15 лет и более, по сравнению с женщинами, которые на чали менструировать до 13 лет, ОР ра ка молочной железы снижен наполо вину. У женщин с поздней менопау зой (54 года и более) ОР увеличен в 4 раза по сравнению с женщинами, у которых менопауза наступила до 47 лет. Роды снижают ОР рака молочной железы. По сравнению с никогда не рожавшей женщиной, у женщины, которая родила одного ребенка, ОР снижен на 50 %. Более того, с увели чением количества беременностей, за вершившихся родами, риск рака мо лочной железы продолжает снижать ся, и у женщины, родившей трех де тей и более, риск на 65 % (ОР равен 0,35) ниже, чем у нерожавших. Ран ние роды также являются фактором, снижающим риск рака молочной же лезы. Так, у женщин, которые родили первого ребенка до 25 лет, ОР рака молочной железы на 35 % ниже, чем у женщин, у которых первые роды были после 35 лет.
Экзогенные гормоны. С точки зре ния возможного канцерогенного рис ка наибольший интерес представляют фармакологические гормональные препараты, получившие значительное распространение в мире — оральные контрацептивы и препараты, приме няемые в качестве гормонозаместительной терапии при менопаузе. Меньшее беспокойство вызывают препараты, которые применяются для профилактики выкидышей, так как их использование в современной меди цинской практике весьма ограничено. Тем не менее первые данные о канце
рогенное™ гормонов получены для диэтилстильбэстрола, нестероидного эстрогена, который широко приме нялся в 40-е годы XX в. для профи лактики выкидышей у беременных женщин. Было показано, что дочери женщин, которые получали во время беременности диэтилстильбэстрол, часто заболевали светлоклеточным ра ком вагины. Частота этого заболева ния достигала максимума в 20 лет, по сле чего риск снижался. Наиболее часто опухоль обнаруживалась у дево чек, матери которых получали диэтил стильбэстрол на ранних стадиях бере менности. Имеются также данные, указывающие на повышенный риск опухолей яичка у мальчиков, матери которых получали этот гормональный препарат.
На основании эпидемиологических данных МАИР признало диэтил стильбэстрол канцерогенным для че ловека и отнесло его в группу 1.
Оральные контрацептивы. Исследо ванию канцерогенного потенциала оральных противозачаточных средств посвящено огромное количество ра бот, при оценке которых необходимо помнить, что в них предметом изуче ния были разные типы оральных кон трацептивов, состав которых менялся в течение всей истории их примене ния. Так называемые последователь ные противозачаточные препараты, поставляющие в организм в течение 14—16 дней достаточно высокие дозы эстрогенов, а в следующие 5—6 дней комбинацию эстрогенов и прогестинов, были изъяты из продажи в конце 70-х годов, так как было показано, что они повышают риск рака эндометрия. Разработанные в последующие годы комбинированные оральные контра цептивы, которые содержат относи тельно низкие дозы эстрогенов и прогестинов, постоянно усовершенство вались в сторону снижения доз входя щих в их состав гормонов и являются в настоящее время наиболее распро страненной формой противозачаточ ных средств в мире. В отличие от по следовательных оральных контрацеп-
56
тивов комбинированные препараты не только не повышают риск рака те ла матки, но и обладают протективным эффектом. Эпидемиологические исследования показали, что у жен щин, которые применяли комбиниро ванные противозачаточные препара ты, на 50 % снижен риск рака тела матки. У женщин, которые получали
этот тип контрацептивов |
более 6— |
10 лет, показатель ОР рака |
эндомет |
рия был равен 0,2. Более того, риск рака этого органа был снижен и у женщин, которые прекратили прием комбинированных контрацептивов 15—20 лет назад.
Результаты многочисленных эпи демиологических исследований пока зали, что применение комбинирован ных контрацептивов снижает риск эпителиальных опухолей яичника. Анализ эпидемиологических исследо ваний свидетельствует о том, что по казатель ОР у женщин, применявших эти препараты, снижен на 40 %, а у женщин с более чем шестилетним анамнезом их применения ОР рака яичника равен 0,3.
Влияние применения комбиниро ванных оральных контрацептивов на риск рака молочной железы, несмотря на большое количество работ, посвя щенных этой теме, остается неясным.
молочной железы у женщин, у кото рых диагноз рака был поставлен до 33 лет и которые начали использовать оральные контрацептивы до первых родов. ОР рака молочной железы у тех женщин, которые получали эти пре параты более 8 лет, был равен 3,5. В этой работе было отмечено, что жен щины, получавшие препараты, содер жащие высокие дозы прогестинов, имеют наиболее высокий риск. Ана логичные результаты получены в ряде эпидемиологических исследований, проведенных в разных странах. На пример, исследование, проведенное в Англии, показало повышение ОР рака молочной железы у женщин в возрас те до 36 лет. Величина ОР в этой по пуляции также зависела от длительно сти использования оральных контра цептивов, у женщин с 8-летним анам незом использования этих препаратов ОР был равен 1,7. В группе женщин, получавших препараты, в которых до за эстрогенов была ниже 50 г, ОР был ниже. Таким образом, можно заклю чить, что прием оральных контрацеп тивов до первых родов скорее всего повышает риск рака молочной же лезы.
Изучению связи между примене нием оральных контрацептивов и ра ком шейки матки посвящено большое
Внескольких эпидемиологических количество эпидемиологических ис
исследованиях, |
в которых изучался |
следований, |
однако другие |
факторы |
||
риск рака молочной железы у жен |
риска, которые могут присутствовать |
|||||
щин, использовавших оральные кон |
у женщин, |
использующих |
оральные |
|||
трацептивы в возрасте более 45 лет, |
контрацептивы в раннем возрасте, ос |
|||||
было выявлено |
статистически |
недос |
ложняют интерпретацию этих резуль |
|||
товерное повышение риска. На осно |
татов. Например, женщины, |
которые |
||||
вании детального анализа этих эпиде |
начинают принимать оральные кон |
|||||
миологических |
исследований |
было |
трацептивы в раннем возрасте, до |
|||
сделано заключение, |
что применение |
первых родов, скорее всего раньше |
||||
оральных контрацептивов в премено- |
начинают активную половую жизнь, |
|||||
паузальный период не связано с рис |
имеют больше партнеров и в связи с |
|||||
ком развития рака молочной железы. |
этим больше подвержены риску быть |
|||||
Однако риск рака молочной железы |
инфицированными вирусом папилло |
|||||
повышен у женщин, которые пользо |
мы человека (ВПЧ), причинная связь |
|||||
вались этими препаратами в раннем |
которого с раком шейки матки дока |
|||||
возрасте и особенно до первых родов. |
зана. В связи с этим результаты эпи |
|||||
Эпидемиологическое |
исследование, |
демиологических исследований, в ко |
||||
проведенное в Лос-Анджелесе, выяви |
торых не учитывались эти "мешаю |
|||||
ло трехкратное повышение риска рака |
щие" факторы, не представляют инте- |
|||||
57
связь между приемом циклических препаратов, которые содержат эстро гены и прогестины, и риском рака яичников, нет.
1.3.4. Потребление алкогольных напитков
Чрезмерное потребление алкоголь ных напитков повышает риск разви тия злокачественных опухолей. На ос новании анализа имеющихся научных данных рабочая группа МАИР при шла к заключению, что алкогольные напитки являются канцерогенным фактором для человека (см. табл. 1.3). Канцерогенность потребления алко гольных напитков для полости рта, глотки, гортани, печени, поджелу дочной и молочной желез можно счи тать доказанной, в то время как в от ношении рака других органов объем доказательств недостаточен.
Втак называемых экологических исследованиях было показано, что по требление алкоголя на душу населе ния коррелирует со смертностью от ряда злокачественных опухолей. Так, например, во Франции была отмечена корреляция между потреблением ал коголя, смертностью от цирроза пече ни и смертностью от рака полости рта, глотки, пищевода и желудка. Аналогичные данные получены в США, где была выявлена статистиче ски достоверная корреляция между потреблением алкогольных напитков на душу населения и смертностью от рака желудка, ободочной и прямой кишки.
ВЯпонии корреляционное иссле дование, которое проводилось в 46 префектурах, выявило связь между
потреблением алкогольных напитков и смертностью от опухолей желудоч но-кишечного тракта. В международ ном исследовании, которое включало 30 стран, выявлена статистически дос товерная корреляция между потребле нием на душу населения алкогольных напитков и первичным раком печени. Корреляция оставалась достоверной после корректировки по заболеваемо
сти гепатитом В. Во Франции, Авст ралии, Англии и Новой Зеландии бы ла отмечена корреляция между дина микой потребления алкогольных на питков и смертностью от рака пище вода, гортани, ободочной и прямой кишки.
Смертность от злокачественных опухолей среди представителей рели гиозных групп, которые воздержива ются от курения и потребления алко гольных напитков, достоверно ниже, чем среди общей популяции. Иссле дования, проведенные среди мормо нов и адвентистов Седьмого дня, про живающих в США, показали, что они значительно реже заболевают раком полости рта, глотки, пищевода, же лудка, ободочной и прямой кишки.
Аналитические эпидемиологиче ские исследования, как проспектив ные, так и ретроспективные, подтвер дили роль потребления алкогольных напитков в канцерогенезе у человека. Статистически достоверное повыше ние ОР развития рака полости рта и глотки было выявлено во всех прове денных когортных и ретроспективных эпидемиологических исследованиях. В некоторых исследованиях ОР был повышен в 10 раз и более. Отмечен синергизм между канцерогенным эф фектом курения и потребления алко голя.
Не вызывает сомнения канцеро генный эффект потребления алко гольных напитков на гортань. Анали тические эпидемиологические иссле дования показали, что ОР возникно вения рака гортани достоверно повы шен у мужчин и женщин, потребляю щих чрезмерное количество алкоголь ных напитков. По данным различных исследований, показатели ОР варьи руют в пределах 15—50 в зависимости от количества потребляемого алкого ля. Во всех эпидемиологических ис следованиях выявлен синергизм влия ния алкоголя и курения на ОР, и этот эффект имеет мультипликативный ха рактер.
Причинную связь между потребле нием алкогольных напитков и раком
59
пищевода также можно считать дока занной. Аналитические эпидемиоло гические исследования выявили зна чительное повышение риска рака пи щевода при чрезмерном потреблении алкоголя. В большинстве исследова ний ОР растет вместе с увеличением количества потребляемого алкоголя и может достигать 10 и более. Курение значительно увеличивает эффект по требления алкоголя на риск рака пи щевода.
Потребление алкоголя приводит и
Связь между потреблением спирт ных напитков и первичным раком пе чени можно считать доказанной. Чрезмерное потребление алкоголя по вышает риск первичного рака печени примерно в 1,5—5 раз. Однако среди населения, в котором распространен другой важный фактор риска для пер вичного рака печени — инфекция ви русом гепатита В и С, влияние по требления алкоголя на риск рака пе чени более выражено. Так, например, среди когорты носителей поверхност
кувеличению риска рака желудка. ного антигена вируса гепатита В ОР,
Влияние алкоголя на процесс канце рогенеза в желудке ограничивается скорее всего кардиальным отделом, хотя в исследовании методом случайконтроль, проведенным в Польше, ОР рака некардиального отдела желудка был значительно повышен у мужчин, которые пили водку натощак.
В исследовании методом случайконтроль, проведенном нами в Моск ве, было выявлено статистически дос товерное повышение ОР рака желудка среди мужчин и женщин, потребляю щих чрезмерное количество алкоголь ных напитков и особенно водки. Кан церогенный эффект потребления крепких алкогольных напитков наи более выражен для кардии. Курение усиливает канцерогенное влияние по требления алкоголя. Результат взаи модействия этих двух факторов на риск рака желудка аддитивный.
Результаты аналитических эпиде миологических исследований о влия нии потребления алкогольных напит ков на риск рака ободочной и прямой кишки противоречивы. В некоторых исследованиях методом случай—кон троль отмечено повышение риска в связи с потреблением крепких спирт ных напитков и пива. Однако в боль шинстве когортных исследований и исследований методом случай—кон троль эти результаты не подтвержда ются. В целом исследования, в кото рых выявлена положительная связь между потреблением спиртных напит ков и риском возникновения опухоли, касаются рака прямой кишки.
связанный с потреблением спиртных напитков, был повышен восьмикрат но, что говорит о синергизме эффекта вируса гепатита В и потребления ал когольных напитков на риск первич ного рака печени.
Положительная и статистически достоверная связь между потреблени ем спиртных напитков и риском рака молочной железы выявлена в более чем двух десятках когортных и ретро спективных эпидемиологических ис следованиях. Результаты мета-анализа 38 эпидемиологических исследований, в которых изучалась связь между по треблением алкоголя и риском рака молочной железы, показали, что ОР рака молочной железы на 30 % выше у женщин, потребляющих спиртные напитки, чем у непьющих.
В исследовании методом случайконтроль, проведенном нами в Моск ве, показано, что риск рака молочной железы статистически достоверно вы ше среди женщин, пьющих спиртные напитки.
Несмотря на впечатляющий объем научной информации, подтверждаю щей канцерогенность потребления спиртных напитков для человека, ме ханизм канцерогенного действия ал коголя все еще неясен.
В экспериментальных исследова ниях этанол как таковой неканцероге нен. Однако этанол играет роль про мотора канцерогенеза в эксперимен тах на мышах, которые получили бенз(а)пирен. Скорее всего подобный эффект спирта можно объяснить его
60
способностью повышать проницае мость мембран клеток. Подобным ме ханизмом канцерогенного действия алкоголя можно объяснить повыше ние риска рака полости рта, глотки, пищевода, желудка, т. е. тех органов, с которыми алкоголь непосредственно соприкасается. Кроме того, этанол, вероятно, воздействует на метаболизм ксенобиотиков и усиливает их повре ждающее действие на ДНК.
1.3.5. Профессиональные канцерогены
Имеющиеся эпидемиологические данные, а также оценка канцероген ного риска для человека профессио нальных факторов, проводимая МАИР, показали, что несколько десятков ве ществ, применяемых в промышленно сти, или промышленных процессов повышают риск развития злокачест венных опухолей и являются канцеро генными для человека. Некоторые из них широко распространены как в высокоиндустриальных странах, так и в странах с сравнительно невысоким уровнем промышленного развития. Кроме того, экспериментальные и эпидемиологические исследования показали, что около 100 веществ, с которыми человек соприкасается в ус ловиях производства, также являются предположительно канцерогенными.
Необходимо отметить, что в тех случаях, когда на основании имею щихся научных данных невозможно выделить конкретное вещество, обла дающее канцерогенным воздействием, принято классифицировать как кан церогенный производственный про цесс, занятость в котором приводит к повышению риска развития злокаче ственных опухолей.
Как видно из табл. 1.4, 4-аминоби- фенил, бензол и 2-нафтиламин повы шают риск развития рака мочевого пузыря. Заболеваемость раком моче вого пузыря повышена среди рабочих, занятых на производстве некоторых красителей — аурамина и фуксина и резины (табл. 1.5). Канцерогенность
Т а б л и ц а |
1.4. |
Химические |
вещества и |
|||
другие |
факторы, |
канцерогенность которых |
||||
для человека доказана (группа 1) |
||||||
Вещество, фак |
Злокачественные опу |
|||||
|
тор |
|
|
холи |
||
4-Аминобифенил |
Рак мочевого пузыря |
|||||
Асбест |
|
|
Мезотелиома плевры и |
|||
|
|
|
перитония, рак легкого |
|||
Афлатоксин |
|
Рак печени |
|
|
||
Бензидин |
|
Рак мочевого |
пузыря |
|||
Бензол |
|
|
Лейкоз, лимфома |
|||
Бериллий и его |
Рак легкого |
|
|
|||
соединения |
|
|
|
|
|
|
Бис(хлорметил) |
Рак легкого |
(мелкокле |
||||
эфир |
|
|
точный) |
|
|
|
Винилхлорид |
Ангиосаркома печени, |
|||||
|
|
|
рак легкого, |
|
опухоли |
|
|
|
|
головного мозга |
|||
Горчичный |
газ |
Рак легкого |
|
|
||
2,3,7,8-тетрахло- |
Все злокачественные |
|||||
родибензопара- |
опухоли, |
рак легкого, |
||||
диоксин |
|
саркома |
мягких тка |
|||
|
|
|
ней, лимфомы |
|||
Ионизирующая |
Лейкозы, другие злока |
|||||
радиация |
|
чественные |
опухоли |
|||
Кадмий и его |
Рак легкого |
|
|
|||
соединения |
|
|
|
|
|
|
Кремний (кри |
Рак легкого |
|
|
|||
сталлический) |
|
|
|
|
||
Мышьяк и его |
Рак легкого, кожи, ан |
|||||
соединения |
|
гиосаркома |
печени |
|||
2-Нафтиламин |
Рак мочевого |
пузыря |
||||
Никель и его со |
Рак легкого, |
пазух носа |
||||
единения |
|
|
|
|
|
|
Радон |
|
|
Рак легкого |
|
|
|
Серная |
кислота |
Рак гортани |
|
|
||
(пары) |
|
|
|
|
|
|
Тальк, |
содержа |
Мезотелиома, рак лег |
||||
щий асбестопо- |
кого |
|
|
|
||
добные |
волокна |
|
|
|
|
|
Ультрафиолето |
Рак кожи, меланома |
|||||
вая радиация |
|
|
|
|
||
Хром и его со |
Рак легкого |
|
|
|||
единения |
|
|
|
|
|
|
Эрионит |
|
Мезотелиома |
|
|||
Этиленоксид |
Гсмобластозы |
|
||||
резиновой промышленности скорее всего связана с использованием на этом производстве 2-нафтиламина. ОР рака мочевого пузыря и легкого повышен у рабочих, занятых в произ водственном процессе коксования уг ля и в алюминиевой промышленно-
61
Т а б л и ц а 1.5. Производственные процессы, канцерогенность которых для человека дока
зана (группа 1)
|
|
Вещество (фактор), ко |
|
|
|
Вид производства |
торое является канце |
Злокачественная опухоль |
|||
|
|
|
рогенным |
|
|
Алюминиевая |
промышленность |
ПАУ |
|
Рак легкого, мочевого пузыря |
|
Газификация угля |
ПАУ |
|
Рак легкого, кожи, мочевого |
||
|
|
|
|
пузыря, мошонки |
|
Коксование угля |
ПАУ |
|
Рак легкого, почки |
||
Литейная промышленность |
ПАУ, |
кремниевая |
Рак легкого |
|
|
|
|
пыль, |
пары металла |
|
|
Производство |
аурамина |
Аурамин |
Рак мочевого |
пузыря |
|
Обувная промышленность |
Бензол |
Лейкоз, лимфома |
|||
Мебельная промышленность |
Древесная пыль |
Рак полости носа |
|||
Производство |
изопропилового |
Диизопропил сульфат, |
Рак носовых |
пазух, легкого, |
|
спирта |
|
изопропиловые масла |
гортани |
|
|
Производство |
фуксина |
Фуксин, ортотолуидин |
Рак мочевого |
пузыря |
|
Резиновая промышленность |
Ароматические ами |
Рак легкого, мочевого пузы |
|||
|
|
ны, растворители |
ря, желудка, толстой кишки, |
||
|
|
|
|
простаты, кожи, гемобластоза |
|
Добыча гематита (подземная) |
Радон |
|
Рак легкого |
|
|
сти. На этих производствах канцеро генное воздействие на человека ока зывают полициклические ароматиче ские углеводороды (ПАУ), с воздейст вием которых связана также повы шенная заболеваемость раком легкого среди рабочих литейных цехов. Необ ходимо отметить, что канцероген ность литейного производства не ог раничивается воздействием ПАУ. Ли тейщики могут быть подвержены воз действию паров хрома, никеля, фор мальдегида, а также кремниевой пы ли. ПАУ чаще являются непосредст венной причиной рака кожи (в том числе и мошонки) у рабочих, контак тирующих с продуктами сгорания угля.
Производственный контакт с бен золом повышает риск лейкоза. Вдыха ние паров серной кислоты приводит к увеличению ОР рака гортани и легко го. Повышение риска ангиосаркомы печени, рака легкого и кожи связано с добычей и выплавкой мышьяка. Кро ме того, мощным канцерогенным дей ствием на печень обладает винилхлорид. Профессиональный контакт с ви-
нилхлоридом повышает риск развития рака легкого, опухолей мозга и лимфогемопоэтической ткани. Производ ственный контакт с асбестом является непосредственной причиной разви тия мезотелиомы плевры и перитония, а также рака легкого.
Соединения бериллия, кадмия и хрома, признанные МАИР канцеро генными для человека, повышают риск рака легкого. Никель и его со единения связаны с повышенным риском рака легкого, носа и носовых пазух. Повышенный риск рака легко го среди шахтеров, добывающих руду, в частности радиоактивную, связан с воздействием повышенных концен траций радона в шахтах. Кроме того, шахтеры подвержены воздействию других соединений, например крем ниевой пыли и мышьяка, которые или сами являются канцерогенными, или могут усиливать канцерогенный эф фект других веществ.
У рабочих, занятых в производстве обуви и деревообрабатывающей про мышленности, значительно повышен риск развития рака носа и носовых
62
пазух. Данных о конкретных канцеро генных веществах, воздействующих на рабочих на этих производствах, нет. По-видимому, пыль, возникающая на рабочем месте в результате обработки кожи и дерева, оказывает раздражаю щее влияние на слизистую оболочку и стимулирует пролиферацию эпителия.
Профессиональный рак кожи опи сан у фермеров и рыбаков и связан с воздействием ультрафиолетовой ра диации. Риск рака кожи повышен у рабочих, контактирующих с продукта ми сгорания угля и минеральными маслами, используемыми при обра ботке металла.
Профессиональная экспозиция различным источникам ионизирую щего излучения связана с увеличени ем риска возникновения лейкозов, опухолей костей, рака легкого, носа и носовых пазух и кожи.
Как было отмечено выше, имеются экспериментальные и эпидемиологи ческие данные о возможной канцеро генное™ для человека многих хими ческих веществ и производств. Одна ко этих данных недостаточно для то го, чтобы отнести к группе 1 вещества или производства, канцерогенность которых для человека можно считать доказанной.
Особенного внимания заслужива ют данные о канцерогенное™ хлорофеноксигербицидов. Результаты эпи демиологических исследований влия ния гербицидов на риск возникнове ния рака не очень убедительны. Одна ко имеются достаточно серьезные данные, указывающие на связь между профессиональным контактом с хлорофеноксигербицидами и риском раз вития саркомы мягких тканей и лим фогранулематоза.
Требует дополнительного эпиде миологического изучения профессио нальный контакт с формальдегидом и искусственными минеральными во локнами, которые широко использу ются в строительстве.
Канцерогенные профессиональ ные факторы редко представлены в виде одного определенного вещества.
Чаще мы имеем дело со сложными смесями, не все составные части ко торых могут быть известны.
Долю случаев рака, причинно свя занных с профессиональным воздей ствием, оценить трудно, но, по имею щимся данным, она составляет около 5 % всех злокачественных новообра зований в развитых странах. Этот про цент может быть выше в регионах с развитой промышленностью: напри мер, заболеваемость раком мочевого пузыря и легкого, связанная с про фессиональным воздействием, может быть очень высока в регионах с разви той промышленностью и слабым ги гиеническим контролем.
Злокачественные новообразования профессионального происхождения, особенно когда причина установлена, более легко поддаются профилактике с помощью соответствующих техноло гических мероприятий, чем те, кото рые связаны с бытовыми факторами.
1.3.6. Загрязнение воздуха
Высокий уровень загрязнения ат мосферного воздуха городов и бли зость места проживания к некоторым промышленным предприятиям могут быть связаны с повышенным риском рака легкого.
К канцерогенным веществам, за грязняющим воздух, относятся ПАУ, хром, бензол, формальдегид, асбест и т. д. В качестве индикатора загрязне ния воздуха ПАУ принят бенз(а)пирен (БП). Основными источниками за грязнения атмосферного воздуха яв ляются предприятия металлургиче ской, коксохимической, нефтеперера батывающей и алюминиевой про мышленности, а также ТЭЦ и автомо бильный транспорт. Уровни ПАУ в атмосферном воздухе значительно превышают ПДК (1 нг/1 м3). Напри мер, металлургический комбинат и коксохимический заводы выбрасыва ют в сутки более 2 кг БП, а нефтепе рерабатывающие заводы — более 3 кг. Концентрации БП в выбросах этих производств чрезвычайно высоки как
63
для рабочей зоны, так и для населен ных мест. Рассеивание выбросов за границу санитарно-защитной зоны создает превышение ПДК для коксо химического производства в 50 раз, для нефтеперерабатывающих заводов в 20 раз. Превышение ПДК распро страняется от предприятий на рас стояние до 10 км. В некоторых рай онах Москвы среднесуточная концен трация БП превышает 20 нг/м\ а ра зовая максимальная концентрация равна 100 нг/м3.
Эпидемиологические данные ука зывают на повышение риска рака лег кого в связи с загрязнением атмо сферного воздуха. В исследовании, проведенном в 26 промышленных го родах СССР, установлено, что заболе ваемость раком легкого среди мужчин коррелирует с показателями загрязне ния атмосферного воздуха. Однако в этом же исследовании выявлено, что корреляция выше для показателей, ха рактеризующих уровни потребления табачных изделий в этих городах. Ис следование методом случай—контроль показало, что ОР рака легкого у неку рящих женщин, связанный с загрязне нием атмосферного воздуха, равен 1,8.
На основании аналитических эпи демиологических исследований мож но сделать вывод, что с учетом риска, вызванного курением, ОР рака легко го, связанный с загрязнением атмо сферного воздуха, не превышает 1,5. В большинстве работ повышение риска рака легкого отмечено только у куря щих.
Наибольшее повышение риска вы явлено у людей, живущих вблизи ме таллургических заводов. У женщин, проживающих рядом с металлургиче ским заводом, риск рака легкого свя зан с уровнем загрязнения воздуха мышьяком. Во всех этих исследовани ях при расчете ОР учитывались факт курения и профессиональная занятость
вметаллургической промышленности. На основании расчетов, сделанных
вэпидемиологических исследованиях, проведенных в Кракове, можно ска зать, что 4,3 % рака легкого у мужчин
и10,5 % у женщин вызвано загрязне нием атмосферного воздуха. В этом же исследовании показано, что 74,7 и 20,6 % случаев рака легкого у мужчин
и47,6 и 8,3 % у женщин вызвано со ответственно курением и влиянием канцерогенных веществ на производ стве. Аналогичные оценки получены и
вряде других эпидемиологических ис следований, проведенных в других странах.
Трудность интерпретации эпиде миологических данных о связи загряз нения атмосферного воздуха с риском возникновения злокачественных опу холей можно объяснить неточностью данных об уровнях канцерогенных ве ществ в воздухе, а также с методиче скими проблемами, связанными с не обходимостью раздельной оценки влияния различных факторов (загряз нение воздуха, курение, профессия). Тем не менее на основании анализа данных эпидемиологических и экспе риментальных исследований можно сделать вывод, что процент злокачест венных опухолей, связанных с загряз нением атмосферного воздуха, не пре вышает 2 % и колеблется в различных странах в пределах 0,1—2 %.
Однако, несмотря на существую щую неопределенность влияния за грязнения атмосферного воздуха на риск злокачественных опухолей, ме ры, направленные на дальнейшее снижение выбросов канцерогенов, вполне оправданы.
1.3.7. Ультрафиолетовое излучение
Данные экспериментальных и эпи демиологических исследований пока зали, что ультрафиолетовое (УФ) из лучение является канцерогенным для человека и приводит к развитию базалиомы, плоскоклеточного рака и ме ланомы кожи.
УФ-излучение является невидимой частью спектра солнечного света с длиной волн 100—400 нанометров (нм). Спектр УФ-радиации условно делится на три части: УФ-С, или так
64
называемые гербицидные УФ-лучи, с длиной волны менее 280 нм; УФ-А- радиация с длиной волны 330— 344 нм, которая вызывает эритему и пигментацию кожи у людей и опухоли у лабораторных животных; УФ-В-ра- диация с длиной волны 280—330 нм. УФ-В-лучи с длиной волны менее 290 нм поглощаются атмосферой и прак тически никогда не достигают земли, небольшая же часть УФ-В-радиации до земли доходит. Именно эта часть спектра УФ-радиации является наи более опасной. Ее влияние на кожу человека, в том числе и канцероген ное, значительно сильнее, чем анало гичный эффект УФ-А-радиации. Изу чение влияния УФ-лучей разной дли ны волны на кожу показало, что наи более эффективно вызывают эритему УФ-лучи длиной волны 297 нм. С уд линением волны "мощность" УФ-лу чей снижается и влияние УФ-радиа ции с длиной волны 325 нм составля ет 0,001 % по сравнению с УФ-лучами с длиной волны 297 нм. Скорее всего УФ-В-радиация играет важную роль и в процессе старения кожи. Показано, что в большинстве плоскоклеточных раков кожи человека в гене-супрессо- ре р53 обнаруживаются мутации, ана логичные мутациям в результате воз действия УФ-В-радиации в экспери ментальных системах. Кроме того, из вестно, что УФ-В-радиация вызывает изменение в иммунологической сис теме, тем самым ингибируя отторже ние трансформированных клеток. С другой стороны, УФ-В-радиация спо собствует повышению в организме уровней витамина D и кальция, что особенно важно для населения с не адекватным питанием.
Основным компонентом атмосфе ры, который защищает нас от чрез мерной УФ-радиации, является озон (03). Озон поглощает УФ-радиацию в стратосфере, пропуская на землю лишь очень небольшое количество УФ-В-лучей. Потеря озона может привести к увеличению количества УФ-В-радиации, достигающей по верхности земли.
Потеря озонового слоя вызвана разными сторонами деятельности че ловека, которая приводит к образова нию в верхних слоях атмосферы оки слов азота, молекул хлора и брома: полетами сверхзвуковых самолетов, использованием азотсодержащих удобрений, а также широким распро странением в быту так называемых фреонов, которые применяются в аэ розольных пульверизаторах, холодиль никах и кондиционерах воздуха. Фреоны являются источником образова ния атомов хлора, которые в страто сфере разрушают озон.
По последним оценкам, которые значительно консервативнее предыду щих, потери озонового слоя с 1969 по 1987 г. составили 3 %. Если не будут приняты соответствующие меры, ог раничивающие выброс в стратосферу веществ, разрушающих озон, то к се редине XXI в. потери озонового слоя могут составить 6 %, а к 2075 г. — 40 %, что приведет к увеличению ко личества УФ-В-радиации, достигаю щей земли. Расчеты, приведенные ко митетом по оценке влияния на эколо гию изменений в стратосфере, пока зывают, что потери 1 % озонового слоя приводят к росту на 2 % уровня УФ-В-радиации в средних широтах. А за этим, по утверждению американ ского агентства по защите окружаю щей среды, должно следовать значи тельное повышение заболеваемости злокачественными опухолями кожи.
Некоторые исследователи связыва ют нынешний рост заболеваемости плоскоклеточным раком и меланомой кожи с повышением уровня УФ-В-ра диации, которая была зафиксирована в некоторых регионах мира — в Кана де и Швейцарии. Однако в целом в мире повышение уровня УФ-В-радиа ции пока не отмечено, а рост заболе ваемости злокачественными опухоля ми кожи скорее всего можно объяс нить тем, что все большее число лю дей из экономически развитых стран отдыхает в жарких странах.
Злокачественные опухоли кожи преобладают среди "белого" населения
5-7908 Д. Г. Заридзе |
65 |
иособенно среди голубоглазых и се роглазых блондинов и рыжеволосых, которые чаще сгорают на солнце и у которых есть склонность к появлению веснушек. Чаще опухоли кожи появ ляются на открытых частях тела.
Вразличных регионах мира отме чается обратная корреляция между за болеваемостью злокачественными опухолями кожи и широтой и поло жительная корреляция с уровнем УФрадиации. Плоскоклеточным раком кожи чаще болеют люди, работающие на открытом воздухе и подвергающие ся длительному воздействию солнеч ных лучей, в то время как меланома кожи встречается чаще среди людей, работающих в помещении, которые, однако, имеют привычку загорать и "сгорать". Риск рака кожи повышен у людей с поражениями кожи, вызван ными солнечными лучами, — кератоз
иэластоз, а также у лиц с такими ге нетическими синдромами, как альби низм, ксеродерма пигментозум. Влия ние УФ-радиации на риск плоскокле точного рака более выражено. В этио логии меланомы наряду с солнечной радиацией очень важную роль играют конституциональные особенности в виде множественных родинок, а осо бенно диспластических невусов.
1.3.8. Ионизирующая радиация
Излучение, которое при взаимо действии с веществом может привести к высвобождению электронов из ато ма и молекул, называется ионизирую щим. Ионизирующие излучения под разделяются на электромагнитные и корпускулярные. К электромагнит ным относятся рентгеновское излуче ние и гамма-излучение. Видимый свет, радиоволны, микроволны, излу чаемые радарными установками, вы соковольтными линиями, сотовыми телефонами и другими промышлен ными и бытовыми установками, — то же электромагнитные излучения, но характеризующиеся большей длиной волны и неспособные ионизировать
атомы и молекулы. Корпускулярное излучение — это заряженные частицы: бета-частицы (электроны), протоны (ядра водорода), альфа-частицы (ядра гелия) и, наконец, нейтроны — не имеющие заряда частицы, которые, однако, могут опосредованно вызы вать ионизацию.
Единицей энергии, поглощенной веществом (тканью), является грей (Гр), равный 100 рад. Биологический эффект радиации зависит не только от поглощенной дозы, но и от энергии, теряемой заряженной частицей на единицу длины ее пробега в веществе, т. е. от показателя линейной передачи энергии (ЛПЭ).
В зависимости от значения ЛПЭ все ионизирующие излучения делят на редко- и плотноионизирующие. К редкоионизирующим относят все электромагнитные излучения, а к плотноионизирующим — протоны, альфа- и бета-частицы. Биологиче ский эффект радиации для 1 Гр варь ирует в зависимости от ЛПЭ и обычно более выражен для высоких значений ЛПЭ, т. е. для плотноионизирующей радиации.
Все виды ионизирующего излуче ния вызывают ионизацию атомов или молекул. Однако облучение объектов разными видами ионизирующей ра диации в равных дозах вызывает раз личные биологические эффекты. По казатель относительной биологиче ской эффективности (ОБЭ) радиации характеризует ее способность вызы вать определенный биологический эффект — гибель клетки, хромосом ные аберрации и т. д. ОБЭ в основ ном определяет ЛПЭ. Для сравнения биологической эффективности раз личных видов излучения пользуются понятием "эквивалентная доза", еди ницей которой является зиверт (Зв). Один зиверт равен 100 бэр (rem). Зи верт представляет собой поглощенную дозу в греях, умноженную на постоян ную, характеризующую тип радиации. Зиверт также используется для оценки радиации смешанного и неидентифицированного типов.
66
Канцерогенность ионизирующей радиации неоднократно была показа на в эпидемиологических исследова ниях, проведенных среди различных групп населения, подвергавшихся об лучению по медицинским показате лям, на рабочем месте, включая ядер ные производства, при испытании атомного оружия, в результате аварии на АЭС и других ядерных установках и, наконец, при атомной бомбарди ровке Хиросимы и Нагасаки. Эти ис следования показали, что ионизирую щая радиация вызывает практически все формы злокачественных опухолей, кроме лимфобластного лейкоза, лим фогранулематоза, рака шейки матки и простаты.
Самым важным источником радиа ции для человека является естествен ная фоновая радиация, представляю щая собой комплекс излучений разно го вида. Его составляющими являются космические лучи (0,27 мЗв/год), ин тенсивность которых колеблется в за висимости от высоты над уровнем мо ря, радиация, излучаемая Землей (0,28 мЗв/год), уровень которой зависит от содержания радиоактивных элементов в почве и горных породах, и радон (2 мЗв/год). К источникам фоновой ра диации относятся и радионуклиды, например калий, которые откладыва ются в организме (0,39 мЗв/год).
Следующую по величине дозу ра диации в течение жизни человек по лучает от источников, применяемых в медицинской практике для диагно стики и лечения (0,53 мЗв/год). Доля радиации, получаемая на рабочем месте и в результате деятельности АЭС, испытания атомного оружия и других искусственных источников, значительно ниже (0,109 мЗв/год). В среднем в год человек получает дозу радиации, равную 1,6 мЗв, исключая радон. Расчеты, основанные на экст раполяции данных исследования эпи демиологов, показали, что воздейст вие в течение жизни 1 мЗв радиации на 100 тыс. населения приведет к воз никновению 65 случаев лейкоза и 495 случаев других форм злокачественных
опухолей. На основании этих расчетов ученые пришли к выводу, что 4—5 % всех злокачественных опухолей чело века причинно связаны с ионизирую щей радиацией. Представленные вы ше оценки будут в будущем скорее всего корректироваться по мере нако пления знаний о влиянии на человека малых доз радиации, так как экстра поляция с больших доз на малые дает недостаточно точные результаты. Од нако уже сейчас можно утверждать, что наиболее эффективным способом снижения влияния радиации на чело века является ограничение использо вания ее для медицинских целей.
Применение ионизирующей радиа ции в медицине. Первые данные о канцерогенности ионизирующей ра диации получены в результате наблю дения за больными, которые часто подвергались ее воздействию. Наблю дение за когортой женщин, больных туберкулезом, показало, что частое флюорографическое обследование, применявшееся для контроля над пневмотораксом — одним из методов лечения туберкулеза, приводило через 10—15 лет после начала лечения к по вышению риска рака молочной желе зы. Риск оставался повышенным в те чение последующих 50 лет. Наиболее высокие показатели риска были за фиксированы у женщин, которым частое флюорографическое обследова ние проводилось в подростковом и детском возрасте. Рост ОР в зависи мости от дозы облучения носил ли нейный характер. Показано, что облу чение молочной железы дозой в 1 Гр увеличивает риск рака этого органа на 60 %, т. е. ОР равен 1,6. Превышение абсолютного риска (ПАР) было равно 10,7 случая на 10" человеко-лет/Гр (ч.- Л./Гр). Повышение риска рака молоч ной железы было зарегистрировано у женщин, которые получали радиоте рапию для лечения послеродового мастита и других неопухолевых забо леваний молочной железы. При этом повышение риска отмечается только через 10 лет после радиотерапии. Не обходимо отметить, что молочная же-
5* |
67 |
леза — один из органов, отличающих ся наибольшей радиочувствительно стью, степень которой зависит от воз раста человека: в период роста и раз вития радиочувствительность выше, чем после 50 лет.
Данных, указывающих на канцеро генный риск, связанный с маммогра фией, нет. Однако с учетом того, что маммография чаще всего рекоменду ется женщинам старше 50 лет, риск скорее всего отсутствует или является минимальным.
Облучение позвоночника для лече ния анкилозирующего спондилеза также связано с повышением риска лейкоза и других злокачественных опухолей. В когорте, состоящей из 14 556 человек, которым проводилась радиотерапия по поводу анкилози рующего спондилеза, была значитель но повышена смертность от лейкозов, рака легкого, пищевода, лимфом, множественной миеломы и опухолей ЦНС. Смертность от лейкозов достиг ла пика через 1—5 лет после лечения, а затем начала снижаться. Пик смерт ности от других форм злокачествен ных опухолей пришелся также на первые пять лет наблюдения, однако повышенный риск сохранялся до 25 лет с момента облучения. ОР лей коза, связанный с облучением в дозе 1 Гр, по данным этого исследования
был |
равен |
1,08, |
а ПАР был |
равен |
0,15 |
на 104 |
ч.-л./Гр. ОР других форм |
||
злокачественных |
образований |
был |
||
выше — 1,14/Гр (ПАР — 4,67 на 104 ч.-л./Гр).
Облучение новорожденных и детей для лечения увеличенной вилочковой железы и миндалин, а также по пово ду tinea capitis, грибкового заболева ния волосистой части головы, приве ло к росту заболеваемости раком щи товидной железы и злокачественными опухолями ЦНС. Повышенный риск сохранялся в течение 40 лет после об лучения. ОР рака щитовидной железы у детей, облученных по поводу увели чения тимуса, был равен 9,9/Гр, а ПАР составляло 2,9 случая на 104 ч.- л./Гр. В когорте же детей с tinea capitis
ОР был равен 31/Гр; ПАР для рака щитовидной железы было равно 13 на 104 ч.-л./Гр. ОР опухолей ЦНС в этой когорте достигал 5.9/Гр; ПАР состави ло 1,14 на 104 опухолей ч.-л./Гр. Уве личение риска лейкоза было показано среди детей, которые получили не большую дозу радиации перинаталь но, в результате диагностической рентгенографии.
Лучевая терапия повышает риск возникновения второй злокачествен ной опухоли у онкологических боль ных. Рост риска лейкоза и лимфомы отмечен у больных, получивших ра диотерапию по поводу рака шейки и тела матки и лимфогранулематоза. Лу чевая терапия в комбинации с химио терапией значительно увеличивает риск лейкоза у больных лимфограну лематозом и раком молочной железы. При этом у больных лимфогранулема тозом значительно возрастает риск ра ка легкого и молочной железы. Луче вая терапия рака молочной железы также повышает риск рака легкого. По-видимому, эта же причина в опре деленной мере способствует и частому развитию рака второй молочной же лезы.
На основании тщательного анализа роли лучевой терапии в возникнове нии вторых опухолей было сделано заключение, что радиотерапия ответ ственна за 5—10 % всех вторых опухо лей. Роль же других факторов, в том числе химиотерапии, гормонального статуса, а в большей степени факто ров образа жизни, которые причинно были связаны и с первыми опухолями (курение, потребление алкоголя, пи тание), представляется более значи мой.
Наблюдение за детьми, облучен ными по поводу ретинобластомы, вы явило значительное повышение у них риска опухолей костей, мягких тка ней, ЦНС и меланомы. Как известно, ретинобластома часто сочетается с пе речисленными выше опухолями, од нако радиация еще больше повышает риск их развития.
Из 900 больных туберкулезом кос-
68
тей и анкилозирующим спондилитом, которых лечили введением радия (224Ra), у 56 была диагностирована остеосаркома. Ожидаемое число остеосарком в данной популяции было 0,3, заболеваемость достигла пика через 6—8 лет после лечения, а затем начала снижаться. Величина ОР не зависела ни от пола, ни от возраста. Кривая до за—эффект имела линейно-квадрати- ческий характер. ПАР было равно 0,8 на 104 ч.-л./Гр. Риск заболеть остеосаркомой в течение жизни в этой группе при дозе облучения в 1 Гр ра вен 2 %.
Риск остеосаркомы в 10 раз ниже в когорте художников, которые наноси ли на циферблаты часов радий (226Ra), облизывая при этом кисточки, чтобы кончик был тоньше. Разница в канце рогенном риске объясняется тем, что 224Ra имеет очень короткий период полураспада (36 дней), выделяет энер гию на поверхности костной ткани, где располагаются эндостициальные клетки, повреждение которых играет критическую роль в процессе костно го канцерогенеза. У 226Ra очень дли тельный период полураспада (600 лет). Он поглощается и равномерно рас пределяется в костной ткани, что при водит к тому, что доза радиации на единицу костной ткани оказывается относительно небольшой.
Использование торотраста (колло идного раствора двуокиси тория) в рентгенодиагностике привело к росту заболеваемости злокачественными опухолями печени и острым миелобластным лейкозом. Гемангиоэндотелиома печени — злокачественная опу холь, которая является типичной только для больных, которым вводил ся торотраст.
Отдаленные результаты облучения населения, связанные со взрывами атомного оружия. Длительное (более 40 лет) наблюдение за когортой из 93 тыс. человек, переживших атомную бомбардировку в Хиросиме и Нагаса ки, выявило, что рост заболеваемости злокачественными опухолями в этой когорте начался с лейкозов, пик забо
леваемости которыми наступил через 10 лет после взрыва. Кривая доза—эф фект определила линейно-квадратиче- скую зависимость частоты заболева ния от полученной дозы облучения. ОР развития лейкоза составил 6,21/ Гр, а ПАР — 2,94 на 104 ч.-л./Гр. Риск, связанный с облучением, был повышен для острого лимфоцитарного, миелоцитарного и хронического миелоцитарного лейкозов, но не для хронического лимфоцитарного и Т- клеточного лейкозов.
Рак щитовидной железы был пер вой солидной опухолью, заболевае мость которой была повышена в ко горте жителей Хиросимы и Нагасаки, подвергшихся атомной бомбардиров ке. Выявлена линейная зависимость риска заболевания от дозы облучения. Отмечается повышение риска для всех гистологических форм, кроме медул лярного рака. На вскрытии установле но повышение частоты маленького, так называемого оккультного (occult) рака. ОР рака щитовидной железы был равен 10,5/Гр, 4,02/Гр и 1,1/Гр для облученных соответственно в воз расте до 10 лет, 10—20 лет и 20 лет и более. Показатели ПАР были равны 4,4, 2,7 и 0,21 на 10* ч.-л./Гр.
Вкогорте значительно повышен риск рака молочной железы, рост за болеваемости которой начался через 10 лет после взрыва, а форма зависи мости частоты возникновения опухо ли от дозы имела линейный характер. Показатель ОР практически не зави сел от возраста облучения. Для этой когорты ОР рака молочной железы
был равен 2,6/Гр, а ПАР составило 6,7 на 104 ч.-л./Гр. Среди населения, пе режившего атомную бомбардировку, отмечалось также повышение риска всех гистологических форм рака лег кого, желудка, толстой кишки, пече ни, яичника, мочевого пузыря и кожи.
Вкогорте, которая наблюдалась тридцать пять лет (1950—1985) и со стояла из 41 791 человека, получивше го при взрыве дозу более 1 сГр, 55,4 % (112 из 209) всех смертей от лейкозов
69
и 10,2 % смертей от других злокачест венных новообразований (585 из 5734) были признаны этиологически свя занными с радиацией. Подсчитан ОР смерти от всех форм солидных опухо лей, который равен 1,63/Гр, а ПАР за 35 лет (1950-1985) равен 29,7 на 104 ч.-л./Гр.
В заключение хочется подчеркнуть, что изучение когорты людей, пере живших атомную бомбардировку, привело к значительному накоплению знаний о радиогенных опухолях чело века. Размер когорты, тщательная оценка доз радиации, длительное на блюдение, меры, направленные на выявление и учет всех заболевших и умерших от злокачественных опухо лей, и другие причины позволили достаточно точно оценить риск воз никновения злокачественных новооб разований в зависимости от дозы и типа радиации, возраста экспозиции и других важных параметров. Однако размера даже этой когорты оказалось недостаточно для прямой оценки рис ков при малых дозах, поэтому для экстраполяции от больших доз к ма лым необходимо использование мате матических моделей доза—эффект, основанных на данных по широкому спектру высоких доз.
Воздействие радиации на рабочем месте. Первая злокачественная опу холь — рак кожи, вызванная радиаци ей, была диагностирована в 1902 г. у рентгенологов. Далее было показано, что у радиологов повышен риск лей козов, миеломной болезни, а также большинства солидных опухолей. Од нако принятие защитных мер значи тельно снизило риск опухолей среди представителей этой профессии.
Получены убедительные данные о канцерогенном эффекте радона среди шахтеров. Эпидемиологическое иссле дование, в которое были включены 11 баз данных из разных стран (США, Швеции, Чехословакии, Китая и т. д.), выявило значительное повыше ние риска рака легкого. Кривая зави симости доза—эффект имела строго линейный характер.
Данные о повышенном риске раз вития злокачественных опухолей сре ди работников различных ядерных ус тановок противоречивы. Большинство эпидемиологических исследований, основанных на наблюдении за этими контингентами, не выявили повыше ния заболеваемости, а в ряде из них отмечен "дефицит" заболевания ра ком, что можно объяснить так назы ваемым эффектом "здорового рабоче го". В некоторых исследованиях пока зано повышение риска лейкоза (кроме хронического и лимфоцитарного) и миеломной болезни и одновременно снижение риска рака легкого и про статы.
Результаты последних исследова ний, в которые были включены пер вичные данные работников различных ядерных предприятий США и Кана ды, говорят скорее о снижении риска рака (ОР равен 0,87) в результате эф фекта "здорового рабочего", чем об его повышении. Нужно подчеркнуть, что доза радиации, полученная работ никами на этих предприятиях, не пре вышала 5 сГр (0,05 Гр). Кооператив ное исследование, в которое были включены данные американских и английских ученых по 76 тыс. работ ников ядерных установок, показало, что только 9 из 3976 случаев злокаче ственных опухолей можно было свя зать с радиацией.
Воздействие радиации, связанное с испытанием ядерного оружия. Среди участников испытания атомного ору жия в Неваде в 1957 г. было обнару жено повышение риска лейкоза. Риск других злокачественных опухолей не был повышен.
В результате выпадения радиоак тивных осадков в 4 атоллах вблизи острова Бикини, которое явилось следствием испытаний атомного ору жия, население этих атоллов получило достаточно высокие дозы радиации. Средняя доза на щитовидную железу для детей в зависимости от возраста была 3—52 Гр, а для взрослых — 1,6— 12 Гр. В течение 60 лет наблюдения у 60 (24 %) из 253 облученных выявле-
70
ны различные узловатые образования щитовидной железы. Рак щитовидной железы диагностирован у 7 из 130 женщин и у 2 из 113 мужчин; 6 из 8 случаев рака щитовидной железы бы ли диагностированы у лиц, получив ших облучение в возрасте до 19 лет. ОР рака щитовидной железы для этой популяции был равен 1,5/Гр.
Эпидемиологическое исследова ние отдаленных последствий выпаде ния радиоактивных осадков в штатах Юта и Невада не выявило повышения риска рака щитовидной железы. В по следующих исследованиях выявлено небольшое повышение заболеваемо сти острым миелобластным лейкозом у лиц, которым во время выпадения радиоактивных осадков было менее 20 лет.
Данные о последствиях испытаний атомного оружия в СССР не публико вались. Однако эпидемиологические исследования заболеваемости детски ми опухолями, проведенное нами в 80-х годах XX в. в четырех областях Казахстана, прилегавших к Семипала тинскому полигону, выявило повыше ние риска (ОР — 1,5) острого лимфобластного лейкоза.
Риск злокачественных опухолей у населения, проживающего вблизи ядерных установок. На основании ис следований, проведенных в Англии, было высказано предположение, что у детей, проживающих по соседству с ядерными установками, повышена за болеваемость лейкозами. Особый ин терес вызвало сообщение, что в по селке, расположенном в 3 км от пред приятия по переработке ядерного топ лива в Селлафильде, лейкозом заболе ло 7 детей. Эта цифра значительно превышала ожидаемое число лейкозов (0,25). Лейкозом заболели только те дети, которые родились в этом посел ке. Необходимо отметить, что среди работников ядерного предприятия в Селлафильде не было обнаружено превышение заболеваемости ни зло качественными опухолями вообще, ни лейкозами в частности. Было высказа но предположение, что причиной лей
козов у детей скорее всего было облу чение отцов до их зачатия, т. е. мута генный эффект радиации на половые клетки. Однако дальнейшие исследо вания не подтвердили этой гипотезы. Оказалось, что часть отцов детей, за болевших лейкозом, были химиками и имели контакт с различными химиче скими веществами, воздействием ко торых также можно объяснить лейкоз у детей. Кроме того, обследование групп населения, проживающих вбли зи других ядерных установок в Вели кобритании и других странах, не под твердило результаты, полученные в Селлафильде.
Отдаленные последствия аварии на Чернобыльской АЭС. Эпидемиологи ческие исследования отдаленных по следствий аварии на ЧАЭС выявили достоверное повышение риска рака щитовидной железы среди детей. Это. повышение частично может быть объ яснено эффектом скрининга. Однако большая часть этих случаев, несо мненно, связана с радиацией. Количе ство детей в Белоруссии, которым в 1986—1989 гг. поставлен диагноз рака щитовидной железы, было менее пя ти, в последующие же 3 года зарегист рировано 114 случаев рака этого орга на. Рост заболеваемости раком щито видной железы был наиболее выражен в Гомельской области, в регионе, жи тели которого получили наиболее вы сокие дозы радиации и, в частности, радиоактивного йода (13'1), экспози ция которому предшествовала экспо зиции другим радиоактивным вещест вам, в частности изотопам цезия. За болеваемость раком щитовидной же лезы выросла и среди детей, прожи вавших в наиболее загрязненных рай онах России и Украины. Небольшой рост частоты этого заболевания был также отмечен в Северной Англии и США. Однако, учитывая то, что пери од полураспада радиоактивного йода очень короткий, вероятность того, что американские и английские дети под верглись воздействию этого радиоак тивного изотопа, попавшего в окру жающую среду в результате аварии на
71
ЧАЭС, крайне низка. Серьезным под тверждением причинной связи между аварией на ЧАЭС и ростом заболевае мости раком щитовидной железы у детей является исследование методом случай—контроль, проведенное в Бе лоруссии, в которое были включены 107 детей, больных раком щитовид ной железы, 107 детей, представляю щих популяционную контрольную группу, и 107 детей, которые участво вали в популяционном скрининге и у которых не было выявлено патологии щитовидной железы. Результаты ана лиза показали, что дети, больные ра ком щитовидной железы, получили значительно более высокие дозы ра диации. ОР рака щитовидной железы был в 6 раз выше у детей, которые по лучили дозу радиации более 1 Гр, по сравнению с теми, кто получил дозу менее 0,3 Гр, и эта разница была ста тистически достоверна. Исследовате ли отмечают, что рак щитовидной же лезы у детей, связанный с аварией на ЧАЭС, имеет исключительно папил лярное гистологическое строение, клиническое течение его более небла гоприятно, опухоли чаще возникают у детей, которые подверглись воздейст вию радиации до пяти лет, и что ла тентный период между воздействием радиации и развитием рака необычай но короток.
Результаты эпидемиологических исследований, в которых изучалась связь межу аварией на ЧАЭС и раком щитовидной железы у взрослых, ме нее убедительны. Тем не менее в двух когортах ликвидаторов, которые на блюдались в Эстонии и России, выяв лено повышение заболеваемости ра ком щитовидной железы по сравне нию с ожидаемой заболеваемостью, основанной на статистике рака щито видной железы в Эстонии и России. Необходимо отметить, что подобное сравнение правомочно, однако, учи тывая проблемы популяционной ста тистики вообще и особенно в России, где, несомненно, имеется недоучет больных злокачественными опухоля ми и прежде всего опухолями щито
видной железы, результаты такого сравнения apriory должны быть поло жительными.
Данные эпидемиологических ис следований не указывают на связь между аварией на ЧАЭС и заболевае мостью лейкозом у детей. Эпидемио логическое исследование, в котором изучалась динамика заболеваемости лейкозами и лимфомами детей в 23 странах, не выявило связи между не большим ростом заболеваемости лей козом, который был отмечен иссле дователями, и радиацией. Аналогич ные исследования на Украине, в Бе лоруссии, Финляндии не выявили роста заболеваемости детским лейко зом. Однако в Швеции получены ре зультаты, на основании которых вы сказано предположение о незначи тельном и статистически недостовер ном росте среди детей заболеваемо сти острым лимфобластным лейко зом. В исследовании, проведенном в Турции, был выявлен рост заболевае мости лейкозом у детей младенче ского возраста, рожденных в 1986— 1987 гг., по сравнению с заболеваемо стью детей того же возраста, рожден ных до 1986 и после 1987 г. Когорта детей, в которой было выявлено по вышение риска лейкоза младенческо го возраста, была apriory признана турецкими исследователями как экс понированная радиацией в результа те аварии на ЧАЭС, хотя причина повышения заболеваемости лейкозом могла быть другая, в первую очередь связанная с влиянием промышлен ных и бытовых химических веществ как на самих детей, так и на их роди телей. Однако нельзя также исклю чить связи роста заболеваемости лей козом с воздействием радиации на плод in utero или в раннем младенче ском возрасте. Статистически досто верное повышение заболеваемости лейкозом в младенческом возрасте было выявлено в так называемой экспонированной когорте, т. е. ко горте 1986—1987 гг. рождения, в Гер мании. Однако при дальнейшем ста тистическом анализе данных с уче-
72
том дозиметрии результаты не были подтверждены.
Исследования динамики заболе ваемости лейкозом взрослого населе ния в наиболее загрязненных регио нах Украины и Белоруссии не выяви ли роста заболеваемости, которую можно было бы объяснить воздейст вием радиации. Однако повышение риска острого лейкоза было отмечено среди ликвидаторов, получивших наи большие дозы радиации.
Риск злокачественных опухолей, связанный с воздействием радона в по мещениях. Радон является источни ком половины всей дозы ионизирую щего излучения, получаемого челове ком. Как было показано выше, у шах теров в результате воздействия радона значительно повышен риск рака лег кого. Однако уровень радона в жилых помещениях значительно ниже, чем в шахтах, и поэтому изучение канцеро генного воздействия радона в поме щениях крайне трудно.
Несколько исследований, прове денных в Швеции, США, Китае, и наше исследование в Москве выявили повышение риска рака легкого у жен щин, связанное с высокими уровнями радона в жилых помещениях. Комби нированный мета-анализ этих иссле дований показал, что ОР рака легкого вследствие воздействия радона в по мещениях равен 1,2, а процент рака легкого, который этиологически свя зан с этим фактором, не превышает 2.
Необходимо подчеркнуть, что вы сокие уровни радона характерны для домов из камня и особенно гранита, а также для первых этажей домов, по строенных в скалистой местности.
7.3.9. Инфекционные
факторы
Вирус гепатита В (HBV). Частота хронической инфицированности HBV колеблется от высокой в странах ЮгоВосточной Азии и Центральной Аф рики, где носителями хронической инфекции являются более 8 % населе ния, до низкой в Европе и Северной
Америке, где частота инфицированно сти не превышает 2 %.
Вэндемичных регионах HBV чаще всего передается перинатально от ма тери к ребенку, а также в раннем дет ском возрасте от ребенка к ребенку. В 70—90 % этих случаев инфицированность не проявляется клинически и приобретает хронический характер.
Вразвитых странах инфекция HBV
восновном распространяется среди взрослых парентерально и половым путем, что приводит к развитию гепа тита, и только в 5—10 % случаев инфицированность приобретает хрони ческий характер.
Основным серологическим марке ром инфицированности HBV является HBsAg, поверхностный антиген. Если HbsAg определяется в сыворотке бо лее 6 мес, это указывает на то, что инфицированность приобрела хрониче ский характер. Имеется выраженная корреляция между инфицированностью населения HBV и заболеваемо стью гепатоцеллюлярным раком.
Результаты более десятка проспек тивных когортных исследований по казали, что хроническая инфицированность HBV в 100 раз и более повы шает риск развития гепатоцеллюлярного рака. Исследования методом слу чай—контроль также выявили связь между серологическими показателями инфицированности HBV и относи тельным риском гепатоцеллюлярного рака. ОР в этих работах колебался в пределах 5—30. Риск возникновения рака значительно выше у мужчин, чем у женщин, что скорее всего связано как с гормональным профилем муж чин, так и с особенностями образа жизни, т. е. потреблением алкоголя и курением.
Учитывая, что заражение HBV ча ще всего происходит перинатально или в раннем детском возрасте, а опу холи в печени у подавляющего боль шинства больных возникают после пятидесяти лет, очевидно, что процесс канцерогенеза в печени протекает в течение длительного латентного пе риода.
73
Т а б л и ц а 1.6. Инфекционные агенты
Канцероген |
Ассоциированные ново |
|
ные вирусы |
образования |
|
|
|
|
ВГ В (HBV) |
Гепатоцеллюлярный |
рак |
ВГ С (HCV) |
Гепатоцеллюлярный |
рак |
ВТЛЧ |
Т-клеточный лейкоз |
|
(HTLV I) |
взрослых |
|
ВПЧ (HPV), |
Рак шейки матки |
|
16-й, 18-й ти |
|
|
пы |
|
|
ВЭБ (EBV) |
Лимфома Беркитта, рак |
|
|
носоглотки, лимфограну |
|
|
лематоз, рак желудка |
|
ВИЧ (HIV), |
Саркома Капоши, лим- |
|
1-й тип |
фомы |
|
КАНЦЕРО |
|
|
Г Е Н Н Ы Е |
|
|
БАКТЕРИИ И |
|
|
ПАРАЗИТЫ |
|
|
Helicobacter |
Рак желудка |
|
pylori |
|
|
Schistosoma |
Рак мочевого пузыря |
|
haematobium |
|
|
Opistorchis |
Холангиоцеллюлярный |
|
viverrini |
рак |
|
|
|
|
На основании анализа существую щих эпидемиологических данных ра бочая группа МАИР пришла к заклю чению, что хроническое носительство вируса гепатита В является канцеро генным для человека — группа 1 (табл. 1.6).
Показано, что афлатоксины, кан церогенность которых для человека доказана, повышают риск развития печеночно-клеточного рака у HBVносителей: они играют роль кофакто ра в канцерогенезе.
Для оценки эффективности вакци ны против гепатита В в профилактике рака печени в Гамбии проводится ин тервенционное контролируемое про спективное исследование. В то же время во многих странах Азии и Аф рики принята практика массовой вак цинации новорожденных. В западных странах рекомендуется тестирование на HbsAg всех беременных женщин, а младенцам, рожденным от женщин с положительным HbsAg, проводится вакцинация.
Вирус гепатита С (HCV). Частота носительства HCV в различных регио нах колеблется от менее чем 1 % в Ев ропе до 1—3 % в странах Ближнего Востока и Азии.
HCV чаще всего передается парен теральным путем. К группе риска в первую очередь относятся наркоманы, больные, которым проводится гемо диализ и частые переливания крови, а также медицинские работники. Пере дача HCV половым путем или перина тально происходит реже. Инфекция HCV обычно приобретает хрониче ский характер и вызывает тяжелый хронический гепатит, а в дальнейшем цирроз.
Тестом инфицированности HCV является выявление в сыворотке кро ви антител к HCV или непосредствен но РНК вируса. Тест-системы второго поколения характеризуются высокой чувствительностью и специфично стью.
Результаты проспективных когорт ных исследований показали, что на личие антител к HCV, т. е. инфицированность HCV, является маркером по вышенного риска печеночно-клеточ ного рака.
Исследования методом случайконтроль показали, что инфицированность HCV повышает риск рака пече ни. В работах, в которых использова лись тест-системы первого поколения, показатель ОР достигал 100, а при ис пользовании тест-системы второго поколения ОР не превышал 52.
На основании анализа существую щих эпидемиологических данных ра бочая группа МАИР по оценке канце рогенного риска пришла к выводу, что хроническое носительство вируса гепатита С является канцерогенным для человека, a HCV отнесен к группе 1 доказанных канцерогенов (см. табл. 1.6).
Для профилактики гепатита С и первичного рака печени, причиной которого является HCV, необходимо получение вакцины против HCV, ра бота над которой ведется в ряде лабо раторий мира.
74
Вирус папилломы человека. Вирус папилломы человека (HPV) чаще все го передается половым путем. Воз можны также перинатальный и ораль ный пути передачи инфекций. Инфи цирование наиболее вероятно на уча стках поврежденного эпителиального покрова. Процент носительства HPV наиболее высок среди сексуально ак тивных молодых людей. Частота ин фицированности одинаково высока среди обоих полов.
Заражение HPV в подавляющем большинстве случаев не приводит к заболеванию и не дает никаких сим птомов. Однако у определенного про цента инфицированных возникают кондиломы и папилломы дыхательных и половых органов, других слизистых оболочек, а также папилломы и боро давки на коже. И только у очень не большого процента HPV-инфициро- ванных развивается предрак и рак шейки матки. Результаты эпидемио логических исследований подтвержда ют, что HPV 16-го и 18-го типов этио логически связаны с раком шейки матки.
Дескриптивные эпидемиологиче ские исследования выявили корреля цию между заболеваемостью раком шейки матки и частотой инфициро ванности HPV. Процент женщин с носительством HPV значительно вы ше среди населения с высокой заболе ваемостью раком шейки матки, чем среди популяции с низкой заболевае мостью.
HPV 16-го и 18-го типов обнару живаются в подавляющем большинст ве случаев рака шейки матки, а также с меньшей частотой при других раках аногенитальной зоны. Необходимо отметить, что применение метода полимеразной цепной реакции (PCR) для выявления вирусного ДНК спо собствовало получению более ста бильных результатов и улучшению выявляемое™ HPV.
Частота выявляемое™ HPV растет параллельно с выраженностью про цесса малигнизации и при интраэпителиальной неоплазии III степени
достигает 70 %. При карциноме in situ, плоскоклеточном раке и аденокарциноме шейки HPV выявляется в 90 % случаев и более. Частота выяв ляемое™ HPV при раке шейки матки практически одинакова во всех регио нах мира. Исследование, проведенное МАИР, в котором были изучены пре параты от более чем 1000 случаев рака шейки матки у женщин из 22 стран, ДНК HPV было обнаружено в 93 % всех случаев, HPV 16-го типа был вы явлен в 50 % всех опухолей, a HPV 18го, 45-го, 15-го типов соответственно
в14, 8 и 5 % всех случаев рака шейки матки. Аналогичные результаты были получены и в других исследованиях, когда использовался метод PCR.
Вкогортных проспективных иссле дованиях изучена роль HPV-инфек- ции на разных стадиях канцерогенеза
вшейке матки. В первом типе когорт ных исследований изучался риск раз вития интраэпителиальной неопла зии шейки матки у здоровых женщин с положительными показателями на HPV. Показано, что у таких женщин достоверно повышен риск развития интраэпителиальной неоплазии. В од ном из исследований, проведенном в США, обследованы на инфицированность HPV 241 женщина, которые в дальнейшем наблюдались и обследо вались в течение двух лет. ОР разви тия интраэпителиальной неоплазии шейки матки с выраженной дисплазией у женщин с положительными ре зультатами на HPV был равен 11, ОР был выше для HPV 16-го и 18-го ти пов, а у женщин с персистирующей инфекцией, т. е. когда HPV-инфици- рованность выявлена в 3 и более по следовательных тестах, ОР равнялся
20.В целом кумулятивная заболевае мость интраэпителиальной неоплази-
ей |
с выраженной дисплазией была |
28 |
% среди женщин с положительны |
ми и 3 % среди женщин с отрицатель ными показателями на HPV.
В другом аналогичном американ ском исследовании 11 тыс. женщин, которые наблюдались в течение 4 лет, кумулятивный риск развития интра-
75
эпителиальной неоплазии с различной выраженностью дисплазии равнялся 60 %.
Инфицированность HPV также яв ляется определяющим фактором про грессии интраэпителиальной неопла зии от слабой до выраженной диспла зии, предрака и рака шейки матки. В исследовании, проведенном в Англии, из 26 женщин, у которых в течение 19—30 мес развилась выраженная дисплазия, у 22 (85 %) при первичном обследовании выявлена инфициро ванность HPV 16-го типа, в то время как из 74 женщин, у которых не была выявлена выраженная дисплазия, толь ко у 17 (23 %) при первичном тестиро вании был выявлен HPV 16-го типа.
Убедительные доказательства роли HPV в этиологии рака шейки матки получены в эпидемиологических ис следованиях методом случай—кон троль. Суммируя результаты этих ис следований, можно сказать, что носи тельство HPV в 10 раз и более повы шает риск рака. В тех работах, где для выявления ДНК HPV использовался метод PCR, показатели ОР, по дан ным большинства исследований, варьируют в пределах 25—100. Наибо лее часто при предраке и раке шейки матки обнаруживаются HPV 16-го и 18-го типов.
Серологические исследования, в которых изучались антитела к различ ным HPV-антигенам и их связь с рис ком рака шейки матки, дали анало гичные результаты, но показатели ОР были ниже.
HPV скорее всего является этиоло гическим фактором и других форм злокачественных опухолей, а частно сти рака вульвы, полового члена и анального рака. Однако, учитывая вы сокую частоту инфицированности на селения HPV и крайнюю редкость этих форм рака, HPV скорее всего не является достаточным фактором, и для развития злокачественной опухо ли необходимы другие дополнитель ные факторы, или кофакторы.
В ряде исследований показана связь между инфицированностью
HPV и раком полости рта и гортани, злокачественными опухолями, основ ной причиной которых является куре ние. В связи с этим возникает необхо димость дальнейшего изучения роли HPV в этиологии рака этих органов. На основании анализа существующих научных данных рабочая группа МАИР пришла к заключению, что HPV 16-го и 18-го типов являются канцерогенными для человека (см. табл. 1.6).
Вирус Эпштейна—Барр. Вирусом Эпштейна—Барр (EBV) инфицирова ны более 90 % взрослого населения. Заражение EBV обычно происходит в раннем детском возрасте и не сопро вождается никакими клиническими проявлениями. Если же человек зара жается EBV, будучи взрослым, то у него развивается инфекционный мононуклеоз. Перенос EBV от человека к человеку происходит через слюну, и носительство вируса приобретает хро нический характер.
Так как большинство взрослого населения инфицированы EBV, обна ружение EBV в опухоли не обязатель но является признаком этиологиче ской связи между вирусом и данной опухолью. Известно, что В-лимфоци- ты являются резервуаром латентной инфекции EBV, поэтому факт обнару жения EBV в В-лимфоцитах, ин фильтрирующих опухолевую ткань, не должен расцениваться с этиологиче ской позиции. В связи с этим для подтверждения этиологической роли EBV для опухолей человека необходи мо учитывать следующие дополни тельные критерии:
1)процент EBV-положительных случаев в изучаемой форме опу холи;
2)процент EBV-содержащих кле ток в одной опухоли;
3)моноклональность EBV, указы вающая на то, что латентная инфекция присутствовала в клетках до размножения злока чественного клона;
4)экспрессия EBV белков.
76
Лимфома Беркитта. Первой зло качественной опухолью, для которой была доказана этиологическая роль EBV, является лимфома Беркитта. Большинство случаев (до 100 %) лимфомы Беркитта в эндемичных регио нах Африки ассоциированы с EBV, в то же время в неэндемичных регионах частота EBV-ассоциированных случа ев колеблется от 20 до 87 %.
Эпидемиологические исследования методом случай—контроль показали, что у больных лимфомой Беркитта значительно выше титры антител к EBV-капсидному (КА) и раннему ан тигенам (РА), чем у здоровых людей, составлявших контрольную группу.
В исследовании, проведенном в Африке, сравнивались титры антител к EBV в сыворотке крови больных с лимфомой Беркитта и контрольной группы. Было показано, что у всех больных детей с лимфомой Беркитта определялись антитела к EBV, а у 87,2 % титры антител были равны или превышали 1/600. В контрольной группе у 18 % детей актитела к EBV вообще не определялись и только у 14 % титры были равны или превы шали 1/160. Статистический анализ этого и других эпидемиологических исследований показал, что носители EBV, у которых определяется высокий титр антител, имеют повышенный риск развития лимфомы Беркитта. ОР
висследованиях, проведенных в Аф рике, достигает 50—60. В проспектив ных когортных исследованиях также было показано, что у детей, у которых
вдальнейшем развилась лимфома Беркитта, титры антител к EBV КА были значительно выше, чем у здоро вых. Каждое двукратное увеличение титра антител приводило к пятикрат ному росту ОР лимфомы Беркитта. В клетках лимфомы Беркитта вирусная ДНК присутствует в моноклональной форме.
Заболевание малярией повышает риск лимфомы Беркитта. На роль ма лярии как кофактора, способствую щего развитию лимфомы Беркитта, указывает схожесть географического
распространения эндемических очагов лимфомы Беркитта и малярии, а так же динамика заболеваемости. Меры, направленные на предупреждение ма лярии с использованием хининопрофилактики, привели к снижению за болеваемости не только малярией, но и лимфомой Беркитта.
Этиологическая роль EBV показа на также для других типов неходжинских лимфом. У больных различными формами иммунодефицита, в том чис ле и у реципиентов трансплантатов и больных с врожденным и приобретен ным иммунодефицитом, заболевание лимфомой практически всегда ассо циировано с EBV.
Особенно часто с EBV ассоцииро ваны редкие формы лимфомы — синоназальная ангиоцентрическая Т- клеточная лимфома.
Лимфогранулематоз (лимфома Ходжкина). Связь между заболевани ем инфекционным мононуклеозом и последующим риском развития лим фогранулематоза установлена как в когортных проспективных исследова ниях, так и в исследованиях методом случай—контроль. Лимфогранулема тоз развивается примерно у 0,1 % больных, перенесших инфекционный мононуклеоз в среднем через 8—10 лет после первого диагноза.
В исследованиях методом случай, в которые были включены более 2 тыс. больных с лимфогранулематозом, бы ло выявлено, что антитела к КА EBV, который является маркером предше ствующей инфекции, определялись с одинаковой частотой как в группе больных с лимфогранулематозом, так и контрольной, однако у больных с лимфогранулематозом титры антител были достоверно выше. В исследова ниях методом случай—контроль, в ко торых изучались антитела к комплексу PA EBV, присутствие которых указы вает на активную репликацию вируса, титры антител также были выше у больных с лимфогранулематозом, чем в контрольной группе. ОР лимфогра нулематоза, связанный с высоким титром (>1/160, >1/640) антител к КА
77
EBV, по результатам 20 исследований методом случай—контроль колеблется от 2 до 17, показатели ОР выше для высоких титров антител к комплексу РА (2—49). Исследование связи между частотой выявляемое™ и татрами ан тител к комплексу ядерных антигенов выявили аналогичную зависимость. Следует особенно отметить результа ты двух исследований, в которых ОР лимфогранулематоза, связанный с вы соким титром (>40; >80) антител к ядерным антигенам, был равен беско нечности (сю).
Результаты когортных проспек тивных исследований с использова нием серологических маркеров ин фицированности EBV подтверждают результаты исследований методом случай—контроль. Моноклональность EBV в опухолевых клетках и экспрессия белка LMP-1 являются подтверждением этиологической свя зи между EBV и лимфогранулемато зом.
Рак носоглотки. В исследованиях методом случай—контроль и в про спективных исследованиях, проведен ных в Юго-Восточной Азии и США, было показано, что частота обнаруже ния антител к КА и PA EBV достовер но выше у больных раком носоглотки, чем у лиц в контрольной группе (ОР равняется 23—32).
Подтверждения этиологической роли EBV для рака носоглотки были получены в результате массового скрининга на носительство EBV в не скольких провинциях Китая и на Тай ване. В одном из них в программу скрининга были включены 148 тыс. здоровых людей, из которых у 3533 (2,4 %) были обнаружены антитела к КА EBV. Дальнейшее клиническое обследование этой группы позволило выявить 55 (1,7 %) случаев рака носо глотки. В аналогичном исследовании, проведенном в другой провинции Ки тая, были обследованы 20 726 здоро вых людей старше 40 лет. У 5,5 % бы ли обнаружены антитела к КА EBV, среди них у 18 (1,6 %) диагностирован назофарингеальный рак.
Результаты молекулярных исследо ваний, в которых показано, что прак тически все случаи недифференциро ванного рака носоглотки являются EBV-положительными, что EBV-no- ложительные опухолевые клетки моноклональны и содержат EBV ДНК и белки, подтверждают роль EBV в этиологии назофарингеального рака.
Однако в этиологии рака носоглот ки важную роль играют и другие фак торы, в первую очередь питание. В странах Юго-Восточной Азии показа на этиологическая роль соленой ры бы, которая наряду с рисом является основным продуктом питания не только для взрослых, но и младенцев. При недостаточной засолке и в ре зультате длительного хранения рыба начинает портиться и в ней образуют ся токсические и канцерогенные ве щества.
Недостаток в питании овощей и фруктов, другие факторы окружаю щей среды вызывают риск рака носо глотки. Профессиональная экспози ция парам формальдегида, пыли и ды му повышает риск рака этой локали зации. Вдыхание дыма в жилище с традиционным китайским очагом, у которого обычно нет никакого дымо отвода, также является доказанным фактором риска назофарингеального рака.
Другие опухоли. EBV с различной частотой обнаруживается в лимфоэпителиальных опухолях различной локализации и в первую очередь лимфомах желудка, EBV обнаружен при аденокарциноме желудка. Во всех слу чаях EBV в опухолевых клетках моноклонален, что говорит о том, что EBV присутствовал в эпителиальных клет ках слизистой оболочки желудка до экспансии, размножения опухолевого клона.
В исследовании методом случайконтроль, проведенном нами в Моск ве, показано, что у больных аденокарци номой желудка, и особенно аденокарциномой кардиального отдела же лудка, значительно выше титры анти тел КА и PA EBV. Высокий татр этих
78
антител значительно (в 10—17 раз) по вышает риск возникновения адено карциномы кардиального отдела же лудка. Однако данных для подтвер ждения роли EBV в этиологии рака желудка недостаточно.
Рабочая группа МАИР по оценке канцерогенного риска пришла к за ключению, что имеется достаточно данных для подтверждения роли EBV в этиологии лимфомы Беркитта, синоназальной ангиоцентрической Т- клеточной лимфомы, лимфомы у больных с иммунодефицитом, лимфо гранулематоза и рака носоглотки. Ра бочая группа отнесла EBV к группе 1, к которой относятся факторы дока занной канцерогенности для человека (см. табл. 1.6).
Вирус Т-клеточного лейкоза взрос лых. Распространенность среди насе ления вируса Т-клеточного лейкоза взрослых (HTLV I) значительно ниже, чем других онкогенных вирусов, и варьирует от 0,2—2 % в регионах с низкой инфицированностью до 2— 15 % в эпидемических регионах. К эпидемическим регионам относятся Япония, Карибские острова, Южная Америка, Ближний Восток и Цен тральная Африка. Более одного мил лиона носителей HTLV I выявлено в Японии, на островах Окинава, Киуши и Шикоку. В других районах Японии инфицированность HTLV I значи тельно ниже. Наиболее крупным ре зервуаром HTLV I инфекции является Африка, где количество зараженных достигает 5—10 млн. В Европе и Аме рике инфицированность HTLV I крайне редка и встречается в основ ном у эмигрантов из стран с высокой инфицированностью. В целом в мире HTLV I инфицированы 15—20 млн человек.
Чаще всего HTLV I передается от матери к ребенку при грудном вскармливании, однако даже в энде мичных регионах носительство HTLV I среди детей встречается чрезвычайно редко. Частота носительства увеличи вается с возрастом и достигает макси мума у людей в 50 лет и старше, она
выше у женщин, чем у мужчин. Из вестны также два других пути переда чи инфекции: половой и гематоген ный. В эпидемических по инфициро ванности HTLV I регионах отмечается высокая заболеваемость Т-клеточным лейкозом взрослых (Adult T-cell leukemia/lymphoma — ATLL). Дескриптив ные эпидемиологические исследова ния выявили кластеры с высокой за болеваемостью ATLL в Японии, Эква ториальной Африке и Центральной Америке, которые совпадали с рай онами, эндемичными по HTLV I. Сероэпидемиологические исследования показали, что 90 % больных ATLL серопозитивны к HTLV I.
ATLL не представлены в офици альных статистических отчетах, по этому данные заболеваемости этой формой лейкоза лимфомы основаны на результатах специальных исследо ваний. Наиболее высокие показатели заболеваемости ATLL зарегистрирова ны в юго-восточной части Японии. Заболеваемость ATLL также всегда высока в Экваториальной Африке, Центральной Америке и Иране. Еди ничные случаи этого заболевания за регистрированы в Европе и США в основном среди эмигрантов из стран с высокой заболеваемостью. Необходи мо учитывать трудности диагностики ATLL, связанные с недоступностью применения серологического тестиро вания для выявления инфицирован ных HTLV 1, а также зачастую край ней остротой и быстротечностью за болевания. Поэтому многие случаи ATLL не диагностируются, соответст венно имеет место значительный не доучет заболеваемости и смертности от ATLL.
Средний возраст больных ATLL в Японии равен 57 годам; в Африке и Центральной Америке ATLL заболе вают на 15 лет раньше. Средний воз раст больных в этих регионах — 40— 45 лет. Случаи заболевания ATLL в детском возрасте зарегистрированы в Южной Америке. Эти особенности возрастного распространения ATLL говорят о том, что существуют некий
79
кофактор или кофакторы, способст вующие развитию этого заболевания у HTLV I-положительных людей, воз можно, их выраженность или степень воздействия на человека различны в разных регионах мира.
В Японии ATLL заболевают 0,6— 1,5 на 1 тыс. HTLV 1-инфицирован- ных в возрасте 40—59 лет. Кумулятив ный риск заболеть ATLL в течение жизни у HTLV I-носителей обоих по лов равен 1—5 %.
Роль HTLV I в этиологии ATLL подтверждена в аналитических эпиде миологических исследованиях. Когортные исследования HTLV I-носи телей, проведенные в Японии, пока зали, что смертность от ATLL среди HTLV I-положительных мужчин рав на 68,1, а среди женщин 35,8 на 100 тыс. населения, в то время как ожи даемая смертность среди HTLV 1-от- рицательных крайне низка.
Исследования методом случайконтроль, проведенные в Японии, на Ямайке, в Бразилии, Тринидаде и Тобаго, показали, что в эпидемичных районах до 100 % всех больных ATLL, HTLV I серопозитивны. Про цент серопозитивных больных ATLL также очень высок (> 90 %) в неэнде мичных регионах, в то время как час тота HTLV I-носительства среди кон трольной популяции не превышает в неэндемичных районах 1 %, а в энде мичных регионах 8 %. Процент HT LV I положительных больных значи тельно ниже при других формах Г- клеточных (3—52 %) и других лимфом (3-25 %).
Инфицированность HTLV I в мла денческом и детском возрасте, при которой вирус скорее всего передается с молоком матери, по-видимому, иг рает важную роль в патогенезе ATLL. Серологическое обследование мате рей, больных ATLL и другими форма ми лимфом, проведенное в Японии, Тринидаде и на Ямайке, показало, что все без исключения матери больных ATLL HTLV I положительны, в то время как среди матерей, больных другими формами лимфом, HTLV I-
инфицированность не превышала
30%.
Врезультате анализа научных дан ных рабочая группа МАИР пришла к заключению, что данных, указываю щих на канцерогенность HTLV I для человека, достаточно и отнесла HTLV I к группе 1 доказанных канцерогенов (см. табл. 1.6).
Вирус иммунодефицита человека (ВИЧ). ВИЧ, как известно, является возбудителем синдрома приобретен ного иммунодефицита человека (СПИД). ВИЧ-инфицированность также связана с повышенным риском саркомы Капоши (СК). Более того, СК является одним из патологических состояний, на основании которых ставится диагноз СПИДа (disease de fining condition).
До начала эпидемии СПИДа СК встречалась достаточно редко и ее бы ло принято считать болезнью пожи лых мужчин восточно- и южноевро пейского происхождения. Однако в дальнейшем был выявлен очаг с высо кой заболеваемостью в Центральной Африке, где эта форма опухоли со ставляла 8 % в структуре заболеваемо сти злокачественными опухолями.
С развитием трансплантологии частота СК в развитых странах не сколько выросла. Среди больных с пе ресаженными органами, которые по лучали иммуносупрессивную тера пию, СК составляет 5 % от всех зло качественных опухолей. С начала 80-х годов XX в., когда, как известно, на чалась эпидемия СПИДа, заболевае мость СК начала резко расти. Изме нилась и возрастная структура боль ных СК.
Заболеваемость наиболее высока у мужчин в возрасте 30—39 лет и 20—29 лет — среди женщин. Неэпидемиче ской формой СК чаще болели мужчи ны, чем женщины, в соотношении 15:1, в то время как при ВИЧ-ассо циированной форме СК соотношение количества мужчин к количеству жен щин значительно ниже — 3:1.
На основании сравнения и анализа записей регистров, злокачественных
80
опухолей и СПИДа (linkage study) был подсчитан показатель стандартизован ного соотношения заболеваемости СК среди больных СПИДом, который оказался равен 97 по результатам ис следования в Иллинойсе и 71 в Кали форнии.
В когортных исследованиях было показано, что среди ВИЧ-инфициро ванных и больных СПИДом заболе ваемость СК значительно выше, чем среди здорового населения. При этом если СК заболевают 10—20 % всех ВИЧ-положительных, то среди муж чин гомо- и бисексуальной ориента ции частота СК достигает 35—50 %. В исследовании методом случай—кон троль, проведенном в Руанде, антите ла к ВИЧ были обнаружены у 11 из 18 больных СК и у 8 из 200 контрольных лиц (ОР равен 35,0).
СК встречается чаще у больных, заразившихся ВИЧ половым путем, и реже у имевших другие пути передачи инфекции. Среди ВИЧ-инфицирован ных чаще болеют молодые мужчины гомо- и бисексуальной ориентации. По данным некоторых исследований, риск СК среди этой группы ВИЧ-ин фицированных в 2000 раз и более вы ше, чем среди здорового населения. Заболеваемость СК среди ВИЧ-инфи цированных неженатых молодых лю дей, по данным исследования, прове денного в США, равна 540:100 000, а среди ВИЧ-инфицированных жена тых мужчин — 25:100 000. Проспек тивное наблюдение за 88 739 больны ми СПИДом из США показало, что у 13 616 (15 %) развилась СК. Процент заболевших был самый высокий (21 %) среди мужчин гомо- и бисексу альной ориентации. В остальных группах частота СК колебалась в пре делах 1—3 %. Аналогичные данные получены в Испании, где среди боль ных СПИДом СК заболели 36 % го мосексуалистов и 2 % наркоманов.
В ряде работ, в которых была сде лана попытка объяснить высокий риск СК среди ВИЧ-инфицирован ных гомо- и гетеросексуальной ориен тации, обнаружено, что риск СК вы
ше у наиболее сексуально активных больных, имеющих большее число сексуальных партнеров и партнеров, больных различными формами вене рических заболеваний. Все это позво ляет предположить, что в этиологии СК играет роль другой, независимый от ВИЧ инфекционный кофактор, пе редающийся половым путем. Среди известных инфекционных агентов в настоящее время рассматривается прежде всего герпес-вирус 8-го типа (HHV8). ВИЧ отнесен МАИР к груп пе 1, т. е. факторам, канцерогенность которых для человека доказана (см. табл. 1.6).
Helicobacter pylori. Инфицирован ность Helicobacter pylori (HP) тесно коррелирует с возрастом и бытовыми условиями жизни: она выше среди бедных слоев и значительно ниже сре ди населения развитых стран. Однако у трети взрослого населения развитых стран выявляются антитела к HP. У большинства носителей HP не вызы вает никаких клинических проявле ний, однако инфицированность мо жет приводить к развитию хрониче ского гастрита и язвы желудка, у очень небольшой части НР-инфици- рованных развивается аденокарцинома или В-клеточная лимфома же лудка.
С начала 90-х годов XX в. было опубликовано более 30 ретроспектив ных сероэпидемиологических иссле дований методом случай—контроль, в которых выявлена связь между титром антител к IgG и риском рака желудка. К сожалению, не во всех этих иссле дованиях проводилась корректировка к таким известным факторам риска рака желудка, как низкий социальноэкономический статус, питание, куре ние и т. д. Мета-анализ 10 проспек тивных когортных исследований, в которых изучались образцы крови, взятые у здоровых людей, у которых впоследствии развился рак желудка, и контрольной группы, которая также представляла членов когорты, пока зал, что у инфицированных HP стати стически достоверно повышен риск
81
6-7908 Д. Г. Заридэе
развития рака желудка с ОР, равным 2,5. Ассоциация наиболее выражена для рака дистального отдела желудка.
Раку желудка обычно предшеству ют атрофический гастрит и кишечная метаплазия, которые в свою очередь развиваются в результате воспалитель ных изменений в слизистой оболочке желудка. Эти изменения тесно связа ны с инфицированностью HP. В ряде исследований показано значительное (до пятикратного) увеличение риска развития атрофического гастрита с кишечной метаплазией у лиц, инфи цированных HP. Кроме того, имеются данные, указывающие на регрессию этого процесса после эрадикации HP.
Механизм канцерогенного дейст вия HP не установлен. HP вызывает воспаление слизистой оболочки же лудка, что влечет за собой увеличение синтеза простагландинов и гиперпро лиферацию клеток, ингибирует апоп тоз. Наиболее сильное повреждающее действие на слизистую оболочку же лудка оказывают цитокинсодержащие линии HP (СаА+). Инфицирован ность этим типом HP, по-видимому, и играет ключевую роль в развитии рака желудка. Эта гипотеза требует под тверждения в сероэпидемиологических исследованиях.
Таким образом, роль HP в этиоло гии рака желудка можно считать дока занной на основании эпидемиологи ческих исследований, которые пока зали, что инфицированность HP по вышает риск рака желудка примерно в два раза, однако не является единст венной причиной заболевания. Вопервых, известно, что ОР развития злокачественных опухолей, связанный с воздействием инфекционных аген тов, обычно значительно выше. Ин фицированность вирусами гепатита В и С, папилломы человека 16-го и 18-го типов и вирусом герпеса челове ка 8-го типа в 20 раз и более повыша ет риск развития соответственно рака печени, шейки матки и СК. Во-вто рых, НР-инфицированность одинако ва среди мужчин и женщин, в то вре мя как раком желудка в два раза чаще
болеют мужчины. Все вышесказанное подтверждает роль питания и других факторов образа жизни — курение и потребление крепких спиртных на питков — в этиологии рака желудка. В одном эпидемиологическом исследо вании рака желудка, проведенном на ми в Москве, выявлено, что инфици рованность HP повышает риск рака желудка только у курящих, а курение повышает риск рака желудка только у мужчин с HP.
Инфицированность HP скорее все го является причиной лимфомы же лудка, которая развивается из лимфоидной ткани слизистой оболочки (mu cosa associated lymphoid tissue — MALT). Это достаточно редкая опу холь, которая составляет не более 5 % всех злокачественных опухолей же лудка. Исследования показали, что более 90 % больных с лимфомой же лудка HP-позитивны и что инфици рованность HP в 3 раза и более повы шает риск развития MALT. Однако ввиду того что в литературе опублико вано небольшое число наблюдений MALT (70 случаев), этиологическую роль HP нельзя считать доказанной. Опубликованы результаты успешной антибиотикотерапии лимфом желуд ка, но по ряду методических причин, в первую очередь отсутствия в этих исследованиях контрольной группы, нельзя считать эффективность анти биотикотерапии при MALT доказан ной.
HP отнесен МАИР к группе 1 до казанных канцерогенов (см. табл. 1.6).
1.3.10.Наследственность
Прогресс в молекулярной биоло гии способствовал открытию наслед ственных генетических дефектов, приводящих к развитию рака. Некото рые наследуемые дефекты повышают риск возникновения рака в 100 раз и более. В ряде случаев вероятность за болевания у носителей этого насле дуемого дефекта достигает 100 % (в таких случаях "наследственность" яв ляется достаточной причиной для воз-
82
никновения рака). Однако подобные генетические дефекты чрезвычайно редки (1 случай на 10 тыс. населения), в связи с чем редки и связанные с ни ми этиологически злокачественные опухоли. Расчеты, произведенные в различных странах, показали, что процент злокачественных опухолей, этиологически связанных с редкими наследственными синдромами, не превышает 0,1—0,5 %. В то же время по признаку предрасположенности к развитию рака, которая определяется генами, ответственными за метабо лизм канцерогенных веществ, их ак тивацию, детоксификацию, репара цию ДНК, население полиморфно. "Неблагоприятный" фенотип может встречаться у 30—50 % населения. Од нако ОР развития рака в связи с "не благоприятным" фенотипом относи тельно невелик и редко бывает выше 2 - 3 .
Молекулярным субстратом наслед ственной формы рака являются унас ледованные от родителей терминаль ные мутации в одном аллеле гена супрессора. Этот дефект поражает все соматические клетки потомков, а му тации во втором аллеле, которые при водят к малигнизации клетки, явля ются приобретенными и чаще всего носят случайный характер. Таким об разом, инактивация рецессивных ге нов супрессоров — непосредственная причина большинства наследственных
40 % всех случаев этой болезни в от личие от большинства опухолей чело века, наследственная форма которых составляет не более 1—2 %. Ретиноб ластома наследуется по аутосомно-до- минантному типу с высокой пенетрацией. У больного ретинобластомой с двусторонним поражением и семей ным анамнезом ретинобластомы бо лезнь всегда носит наследственный характер. Кроме того, 10 % ретинобластом с односторонним поражением и без заболевания в семейном анамне зе могут быть наследственными и их возникновение связано с новыми тер минальными мутациями. Носители гена ретинобластомы (Rb) часто стра дают умственной отсталостью, микро цефалией и другими врожденными уродствами.
Исследования последних лет пока зали, что Rb инактивирован и в опу холевых клетках ненаследственных, спорадических ретинобластом, а так же при мягкотканой остеосаркоме, которые часто встречаются у больных с наследственной ретинобластомой. Кроме того, инактивация Rb обнару жена при раке толстой кишки и мо лочной железы, что говорит о важном значении этого гена в этиологии зло качественных опухолей человека.
Другой опухолью детского возрас та, которая характеризуется семейным распространением, является опухоль Вильмса. У больных детей она часто
исемейных форм рака. Пожалуй, сочетается со спонтанной аниридией
единственным исключением является наследственный опухолевый синдром множественной эндокринной неопла зии, причиной которого считают тер минальные мутации в онкогене RET.
К наиболее хорошо изученным на следственным злокачественным опу холям и наследственным синдромам относятся ретинобластома, опухоль Вильмса, семейный аденоматозный полипоз толстой кишки, синдром Линча и синдром Ли—Фраумени.
Ретинобластома встречается очень редко, заболеваемость не превышает 3,5 на 1 млн детей. Наследственная форма ретинобластомы составляет до
и врожденными пороками мочеполо вой системы. Однако нужно отметить, что семейная форма опухоли Вильмса встречается относительно редко. В ли тературе описано менее 100 семей с историей опухоли Вильмса, и в ряде этих семей было обнаружено лишь два случая заболевания этой формой опу холи.
Молекулярные исследования по зволили выделить ген-супрессор опу холи Вильмса — WT1. Этот ген повре жден или отсутствует у больных с опу холью Вильмса, которые также стра дают аниридией или имеют врожден ные пороки мочеполовых органов.
6* |
83 |
|
Скорее всего ген WT1 не является единственным маркером семейной (наследственной) опухоли Вильмса.
Семейный аденоматозный полипоз толстой кишки во многие десятки раз повышает риск возникновения рака ободочной и прямой кишки. Этот синдром часто сочетается с умствен ной отсталостью и множественными пороками развития. Проведенные мо лекулярные исследования показали, что причиной развития этого наслед ственного синдрома является ген-су- прессор АРС (adenomatous polyposis coli), который находится на хромосо ме 5q. Этот ген обнаруживается прак тически без исключения во всех семь ях с аденоматозным полипозом тол стой кишки и синдромом Гарднера. Рак толстой кишки развивается прак тически у всех больных с аденоматоз ным полипозом.
Описаны также наследственные, се мейные формы первично-множествен ных опухолей. Синдром Линча, или синдром множественных аденокарцином, характеризуется семейным рас пространением первично-множествен ных аденокарцином ободочной кишки, молочной железы, эндометрия, яични ка, поджелудочной железы, желудка, а также лейкоза и опухолей мозга. На следуется синдром по аутосомно-доми- нантному типу. Однако поиски гена, предрасполагающего к этому синдро му, пока к успеху не привели.
Значительно лучше изучен син дром Ли—Фраумени, причиной кото рого является наследуемая мутация в одном аллеле гена-супрессора р53. В семьях с этим синдромом повышен риск раннего рака молочной железы и детских опухолей: мягкотканой сарко мы, острого лейкоза, опухолей мозга, надпочечников, возможно, и других опухолей.
Риск рака молочной железы в 2—3 раза выше у женщин, родственницы которых болели раком молочной же лезы. Повышенный риск рака молоч ной железы передается от родителей детям. В этих семьях раком молочной железы часто болеют и мужчины. На
следственный рак молочной железы чаще диагностируется у молодых жен щин репродуктивного возраста. Моле кулярные исследования показали, что наследственный рак молочной железы у молодых женщин этиологически связан с геном BRCA-1 и BRCA-2. На следуемая мутация в гене-супрессоре BRCA-2 ведет к предрасположенности раку молочной железы и у мужчин. Ген BRCA-1 ответствен и за семейный рак яичника. Эпидемиологические исследования показали, что у кровных родственников, больных раком яич ника, в 3 раза повышен риск заболе вания раком этого органа.
К наследственным дефектам, пред располагающим к возникновению злокачественных опухолей, относятся те, которые связаны с репарацией ДНК, они крайне редки. Среди них синдром атаксии-телеангиэктазии, который встречается у 1 % населения и при котором значительно повышен риск рака молочной железы.
Пигментная ксеродерма также яв ляется результатом дефекта репарации ДНК, поврежденной ультрафиолето выми лучами. У таких больных очень высок риск рака кожи, а также у лю дей с альбинизмом, светловолосых блондинов.
На риск возникновения злокачест венных опухолей влияет индивидуаль ная способность организма метаболизировать, т. е. активировать или детоксифицировать попадающие в организм канцерогенные вещества. Наиболее изученными ферментами, влияющими на метаболизм канцерогенных ве ществ, являются ферменты, кодируе мые генами семейства цитохрома Р450, которые участвуют в метаболизме ряда канцерогенных веществ: ПАУ, гетеро циклических аминов, ариламинов, нитрозоаминов, афлатоксинов, стерои дов и т. д. Полициклические аромати ческие углеводы в организме метаболизируются ферментами I фазы системы цитохрома Р450 в более реактивные (канцерогенные) вещества. Например, ген CYP1A1 экспрессируется в легком и катаболизирует ПАУ и ариламины, и
84
от уровня его экспрессии зависит риск рака легкого. В то же время ген CYP1A2 экспрессируется в печени и в большей степени влияет на метаболизм ариламинов. Ген CYP2D6 кодирует фермент, участвующий в метаболизме табакспецифических нитрозосоединений, и повышает активность ННК, со ответственно у людей с нуль-феноти пом по этому гену понижен риск раз вития рака легкого, связанного с куре нием.
Канцерогенные вещества могут детоксифицироваться и удаляться из орга низма с помощью ферментов II фазы цитохрома Р450. Таким образом, канце рогенный эффекг ПАУ или других ве ществ является результатом взаимодей ствия между метаболическими процес сами, ведущими к активации или деток сификации канцерогенных веществ, а также индивидуальной способности ор ганизма репарировать ДНК.
Рекомендуемая литература
Заридзе Д. Г. Молекулярная эпидемиоло гия // Арх. пат. — 1997. — Т. 58, № 1.
-С. 65 - 70 .
Заридзе Д. /'. Эпидемиология детских лей
козов |
// Арх. пат. — 1997. — Т. 59, |
№ 5. |
- С. 6 5 - 7 0 . |
Заридзе Д. Г. Эпидемиология и этиология злокачественных опухолей // Вестн. РАМН. - 2001. - № 9. - С. 6 - 1 4 .
Заридзе Д. Г., Земляная Г. М. Риск рака легкого у некурящих женщин в связи с загрязнением атмосферного воздуха // Экспер. онкол. — 1994. — № 16. — С. 132-1356.
Заридзе Д. Г., Ли Н. П., Мень Т. X. Заболе ваемость злокачественными опухолями детей в четырех областях Казахстана, прилегающих к Семипалатинскому по лигону // Мед. радиол. — 1993. —
№ 11. |
- С. 2 4 - 2 8 . |
|
|
|
Заридзе Д. Г., Мень Т. X. Приоритетные на |
||||
правления |
противораковой |
борьбы // |
||
Росс, онкол. журн. — 2001. — № 5. — |
||||
С. 5 - 14 . |
|
|
|
|
Заридзе Д. |
Г, |
Некрасова |
Л. И., |
Мень Т. X. |
Факторы риска рака мочевого пузыря // |
||||
Вопр. онкол. — 1992. — Т. 38. — |
||||
С. 1066-1073. |
|
|
||
Заридзе Д. |
Г., |
Филиппенко |
В. В. |
Эпидемио |
логия |
рака толстой |
кишки: |
результаты |
|
исследования методом случай—кон троль // Вопр. онкол. — 1991. — Т. 37, № 2. - С. 152-158.
Andrieu N., Prevost Т., Lifanova Y. et at. Varia tion in the interaction between familial and reproductive factors on the risk of breast cancer according to age, menopausal status, and degree of familiarity // Int. Epidem. — 2000. - Vol. 29. - P. 214-223.
Boyle |
P., Zaridze D. |
Risk |
factors |
for |
prostate |
and testicular cancer // Europ. J. Cancer. |
|||||
- |
1993. - Vol. 29A. - P. 1048-1055. |
||||
Cancer Epidemiology and Prevention / Eds. D. |
|||||
Schottenfeld, J. F. Fraumeni. — New York |
|||||
- |
Oxford: Oxford University Press, 1996. |
||||
Cancer |
Prevention |
and |
Control |
/ |
Eds. P. |
Gneenwald, B. S. Kramer, D. L. Weed. — New York — Basel — Hong Kong: Marcel Dekker, 1996.
Zaridze D., Arkadieva M., Day N., Duffy S.
Risk of acute nonlymphocitic leukemia in patients with cancer treated with chemo-
and radiotherapy |
// |
Brit. |
J. Cancer. — |
|||
1992. - Vol. 67. - P. 347-350. |
||||||
Zaridze |
D., |
Mukeria A., |
Duffy |
S. |
Risk factors |
|
of malignant melanoma // |
Int. J. Cancer. |
|||||
- |
1992. |
- Vol. 52. |
- P. 159 - 161 . |
|||
Zaridze |
D., |
Evstifeeva |
T. |
Nass |
use, cigarette |
|
smoking and alcohol consumption and the risk of oral and oesophageal cancer // Oral oncology, Europ. J. Cancer. — 1992. —
Vol. |
28B. - P. 2 9 - 3 5 . |
Zaridze |
D., Kushlinskii K., Moore J. W. et al. |
Endogenous plasma sex Hormones in preand postmenopausal women with breast
cancer: |
results from a case-control study |
|
in Moscow // |
Europ. J. Prev. — 1992. — |
|
Vol. 1. |
- P. |
225-230. |
Zaridze D., Lifanova Y., Maximovitch D. Diet, alcohol consumption and reproductive factors in the case-control study of breast cancer in Moscow // Int. J. Cancer. — 1991. - Vol. 48. - P. 493 - 501 .
Zaridze D., Maximovitch D., Borisova E. Alco hol consumption, smoking and risk of gas tric cancer: case-control study in Moscow, Russia // Cancer Causes Control. — 2000.
-Vol. 11. — P. 363 - 371 .
Zaridze |
D., Maximovitch D., Zemlyanaya G. et |
||||||
al. |
Exposure to |
environmental tobacco |
|||||
smoke and risk of lung cancer in non |
|||||||
smoking women from Moscow, Russia // |
|||||||
Int. |
J. |
Cancer. |
— |
1998. — |
Vol. 75. |
- |
|
P. 335-338. |
|
|
|
|
|
||
Zaridze |
D., |
Bulbulyan |
M., Changina O., Mar- |
||||
garyan A. |
Cohorty |
studies |
of |
chloroprene |
|||
exposed workers in Russia // Chemico-Bi- |
|||||||
ological |
Interaction. |
— |
2001. |
— |
|||
Vol. |
135-136. - |
P. 487 - 503 . |
|
|
|||
85
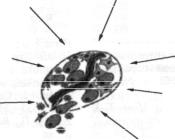
Г л а в а 2
ОСНОВНЫЕ СВОЙСТВА НЕОПЛАСТИЧЕСКОЙ КЛЕТКИ
ИБАЗОВЫЕ МЕХАНИЗМЫ ИХ ВОЗНИКНОВЕНИЯ
Б.П. Копнин
2.1. Характерные признаки опухолевой клетки
Злокачественные новообразования возникают в результате неограничен ной пролиферации клеточного клона, выходящего за пределы собственной ткани и способного к росту на терри ториях других тканей. При этом в си лу высокой генетической изменчиво сти и селекции, происходящей под давлением со стороны организма, в популяции клеток такого клона по стоянно возникают и отбираются все более и более автономные и агрессив ные субклоны, что обозначается тер мином "опухолевая прогрессия". В ре зультате довольно длительной эволю ции неопластического клона форми руется опухоль, способная убить орга низм. В последнее десятилетие был достигнут значительный прогресс как в идентификации генов, нарушения
функции которых ведут к развитию новообразований, так и в выяснении роли белковых продуктов таких генов в физиологии клетки. Все это позво лило выделить ряд важнейших свойств, приобретение которых предо пределяет способность клетки образо вывать злокачественную опухоль (схе ма 2.1).
Во-первых, это пониженная по требность во внешних сигналах для инициации и поддержания клеточной пролиферации — так называемая са
модостаточность в пролиферативных сигналах. Данное положение может быть проиллюстрировано двумя при мерами (схема 2.2). При культивиро вании in vitro большинство типов нор мальных клеток размножается лишь при условии, если питательная среда содержит 10—20 % сыворотки, т. е. при довольно значительном содержа нии в ней различных ростовых факто-
Схема 2.1. Важнейшие свойства неопластической клетки, при
обретаемые в ходе опухолевой прогрессии и обеспечивающие зло качественный рост (объяснение в тексте)
Нечувствительность |
|
Отсутствие репликативного |
к ростсупрессирующим |
|
старения |
сигналам |
|
(иммортализация) |
Самодостаточность |
|
Ослабление |
в пролиферативных |
|
индукции |
сигналах |
|
апоптоза |
Генетическая |
|
Стимуляция |
нестабильность |
|
неоангиогенеза |
Блокирование |
/ |
Изменения |
клеточной |
|
морфологии/локомоции; |
дифференцировки |
|
инвазия и метастазирование |
86

С х е м а 2.2. Характерные признаки |
неопластических клеток i n |
vitro (объяснение в тексте) |
|
Нормальные клетки |
Опухолевые клетки |
Размножения нет |
Пролиферация |
Пролиферация |
|
а |
|
Нормальные клетки |
Опухолевые клетки |
|
Размножения нет |
Пролиферация |
Пролиферация |
|
б |
|
Нормальные клетки |
|
Опухолевые клетки |
Остановка размножения |
Продолжение пролиферации |
в
а — пониженная зависимость от факторов роста; б — способность раз множаться без прикрепления к внеклеточному матриксу; в — отсутствие контактного торможения размножения
ров (схема 2.2, а). Связывание росто |
ки и сотни раз меньшем, чем необхо |
||
вых факторов со своими рецепторами |
димо |
для стимуляции размножения |
|
инициирует передачу сигналов внутри |
нормальных |
клеток. Такая понижен |
|
клетки, приводящую к репликации |
ная потребность в растворимых росто |
||
ДНК и делению клетки. Оказалось, |
вых факторах достигается изменения |
||
что многие типы опухолевых клеток |
ми в системах внутриклеточной сиг |
||
способны размножаться в среде с 1 % |
нализации, |
которые либо вызывают |
|
и даже 0,1 % сыворотки, т. е. при со |
секрецию необходимых факторов рос |
||
держании ростовых факторов в десят |
та |
самими |
трансформированными |
87
клетками, либо резко увеличивают ко личество рецепторов для необходимых факторов роста, либо запускают в от сутствие ростового фактора каскад со бытий, аналогичный тому, который в норме инициируется связыванием ростового фактора со своим рецепто ром.
Другим примером пониженной по требности неопластических клеток во внешних пролиферативных сигналах является их так называемая независи мость от субстрата (anchorage-inde pendence) — схема 2.2, б. Большинст во типов нормальных клеток способ ны размножаться лишь при условии их прикрепления к определенному внеклеточному матриксу. Например, фибробласты начинают делиться при взаимодействии с фибронектином. В ином случае пролиферативный сти мул, исходящий от растворимых рос товых факторов, не вызывает полно ценного каскада передачи внутрикле точных сигналов, необходимого для стимуляции размножения клеток. Многие типы опухолевых клеток в от личие от их нормальных предшест венников способны пролиферировать, не прикрепляясь к субстрату, напри мер, в полужидкой среде. Эти два примера показывают, что неопласти ческие клетки приобретают способ ность генерировать внутри себя пролиферативные сигналы, в норме исхо дящие от внешних стимулов.
Вторым важнейшим приобретен ным свойством неопластических кле ток является их пониженная чувстви
тельность к ростингибирующим сиг налам. Как известно, в организме су ществует множество антипролиферативных сигналов, поддерживающих определенное число клеток в каждой из тканей. Такие сигналы генерируют ся как секретируемыми растворимы ми факторами (цитокинами), так и взаимодействиями клеток с внекле точным матриксом и друг с другом. Классическим примером здесь являет ся так называемое контактное тормо жение размножения клеток в культу рах in vitro (схема 2.2, в).
Нормальные клетки, например фибробласты, размножаются до тех пор, пока не возникнет плотный мо нослой и не установятся межклеточ ные контакты. В отличие от этого трансформированные клетки при воз никновении межклеточных контактов не останавливают свою пролифера цию, а продолжают делиться, напол зать друг на друга и образовывать оча ги многослойного роста.
Наряду с этим опухолевые клетки, как правило, значительно менее чув ствительны к действию ростингибирующих цитокинов, факторов специ фического и неспецифического про тивоопухолевого иммунитета, а кроме того, не останавливают свою проли ферацию при ДНК-повреждающих воздействиях или неблагоприятных условиях — недостатке пула нуклеоти дов, гипоксии и т. д.
Еще одним важнейшим свойством опухолевых клеток является отсутст вие репликативного старения, или приобретение бессмертия (имморта лизация). Как известно, существует механизм, ограничивающий число де лений большинства типов зрелых кле ток человека. Так, в культурах челове ческих фибробластов in vitro после 60—80 делений (так называемое число Хейфлика) наблюдается необратимая остановка размножения клеток и их постепенная гибель. Между тем, что бы образовать из одной клетки-родо начальницы сначала опухоль, а затем и метастазы, в условиях жесткого дав ления со стороны организма, когда многие опухолевые клетки погибают, может потребоваться большее число делений. И, действительно, в опухоле вых клетках наблюдается нарушение работы такого "счетно-ограничитель ного" механизма контроля реплика ции.
Следующим важным свойством не опластических клеток является ослаб ление в них индукции апоптоза. Апоп тоз представляет собой активный ме ханизм клеточного самоубийства, поддерживающий в организме опреде ленное число клеток и, кроме того,
88
защищающий его от накопления ано мальных клеточных вариантов. Он вызывается как физиологическими сигналами (связыванием специфиче ских киллерных цитокинов со своими рецепторами), так и различными внутриклеточными повреждениями или неблагоприятными условиями, в частности нарушениями структуры ДНК, нехваткой ростовых факторов, гипоксией и т. д. Уход от апоптоза резко повышает жизнеспособность неопластической клетки, делает ее ме нее чувствительной к факторам про тивоопухолевого иммунитета и тера певтическим воздействиям.
К приобретенным свойствам опу холевых клеток принадлежит и их спо
собность стимулировать неоангиогенез, т. е. формировать новые крове носные и лимфатические сосуды из эндотелиальных клеток предсуществующих окружающих мелких сосудов. Это необходимое условие для даль нейшего роста опухолевого узелка, достигшего в диаметре 2—4 мм. В ином случае клетки в центре опухоли, не получая кислорода и питательных веществ, будут погибать.
Важнейшим свойством опухолевых клеток являются изменения морфоло гии и движения клеток. В основе мор фологических нарушений лежат взаи мосвязанные между собой изменения цитоскелета, адгезионных взаимодей ствий клеток друг с другом и с вне клеточным матриксом. Они выража ются в нарушении формирования фо кальных контактов и ухудшении при крепления клеток к внеклеточному матриксу, дезорганизации системы актиновых микрофиламентов, что приводит к изменениям активности псевдоподий и подвижности клеток (см. раздел 3.3). В целом наблюдаемая картина напоминает изменения, воз никающие в норматьных клетках при действии мотогенных цитокинов — факторов, стимулирующих миграцию клеток. Однако так называемый локо моторный фенотип в неопластических клетках, как правило, сильно утриро ван, что позволяет различать по мор
фологии опухолевую клетку от движу щейся нормальной. Необходимо под черкнуть, что именно эти нарушения вместе с некоторыми другими свойст вами, в частности способностью секретировать протеолитические энзимы, предопределяют приобретение не опластическими клетками двух свойств, лежащих в основе злокачест венного роста: способность к инвазии,
т. е. проникновению в окружающие здоровые ткани, и сопряженную с ней
способность к метастазированию —
образованию вторичных очагов опухо левого роста. Метастазирование — наиболее опасное проявление опухо левой прогрессии, являющееся основ ной причиной смерти онкологических больных. Чтобы дать метастаз, клетка должна приобрести ряд свойств: спо собность проникать в глубину окру жающих нормальных тканей, в том числе в кровеносные или лимфатиче ские сосуды, выживать после попада ния в сосуды, а затем выходить из них
иразмножаться в несвойственном для данного типа клеток микроокруже нии, давая новый очаг опухолевого роста. Таким образом, способность к метастазированию складывается из комплекса более простых признаков, таких как приобретение локомоторно го фенотипа, возможность стимулиро вать образование новых кровеносных
илимфатических сосудов, создавая тем самым пути эвакуации опухоле вых клеток из первичного очага, неза висимость от субстрата, подавление апоптоза и т. д. Появление каждого из этих свойств увеличивает метастатиче ский потенциал клетки.
Для многих опухолевых клеток ха рактерны и нарушения клеточной дифференцировки, т. е. образования спе циализированных типов клеток, син тезирующих специфические белки. Особенно ярко это проявляется в гемобластозах, новообразованиях из кроветворных тканей, при которых их клетки оказываются как бы заморо женными на той или иной стадии со зревания. Общепринятым является представление, согласно которому
89
С х е м а 2.3. Модели, объясняющие происхождение новообразо
ваний из незрелых клеток определенной стадии дифференцировки, в которых либо сохранена (внизу слева), либо блокирована (внизу справа) способность к дальнейшему созреванию
Стволовая клетка
Амплифицирующие, или коммитированные, незрелые клетки
Терминальнодифференцированные клетки
Нормальная дифференцировка
Хронический |
миелоидный |
лейкоз, |
Большинство |
лейкозов, |
|
плоскоклеточный |
рак |
кожи, |
ряд солидных |
опухолей |
|
другие |
солидные |
опухоли |
|
|
|
меньшая зрелость лейкозных клеток является не следствием дедифференцировки зрелых клеток, претерпевших неопластическую трансформацию, а отражает их происхождение из незре лых клеток, в которых блокированы процессы дальнейшей дифференцировки. Следует заметить, однако, что это свойство не является универсаль ным: во многих типах опухолей на блюдается сохранение способности к дифференцировке, причем в отличие от лейкозов созревание клеток не пре пятствует приобретению злокачест венного фенотипа. Примерами этого могут служить плоскоклеточный ороговевающий рак кожи и высокодифференцированные аденокарциномы
толстой кишки, происходящие из не зрелых клеток, которые сначала не сколько раз делятся, а затем диффе ренцируются (схема 2.3).
Вход в цикл и продвижение по не му определяется последовательной активацией различных циклинзависимых киназ (cdk). Повышение их активности инициируется сигналами от рецепторов ростовых факторов и интегринов (рецепторов белков вне клеточного матрикса), вызывающими сложный каскад событий, приводящих к активации группы МАР-киназ — ключевых регуляторов cdk. Цен тральную роль в негативной регуля ции размножения клеток играют ин гибиторы циклинзависимых киназ
90
(CKI) — белки семейств Ink4 и Cip/ |
2.2. Механизмы |
|
|
|
|
|
|
|||||||||||||||||||||
Kip. |
|
|
|
|
|
|
|
|
|
|
|
|
|
возникновения характерных |
||||||||||||||
Происхождение |
из |
незрелых |
кле |
свойств неопластической |
|
|||||||||||||||||||||||
ток не противоречит представлению о |
клетки |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
том, что опухолевые клетки в ходе |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
прогрессии |
могут |
претерпевать |
опре |
Приобретение совокупности |
пере |
|||||||||||||||||||||||
деленную |
дедифференцировку, |
утра |
численных свойств связано с наруше |
|||||||||||||||||||||||||
чивая в первую очередь те дифферен- |
ниями в сигнальных путях, контроли |
|||||||||||||||||||||||||||
цировочные белки, |
отсутствие |
кото |
рующих реакцию клетки на экзоген |
|||||||||||||||||||||||||
рых дает клеткам селективные пре |
ные и эндогенные регулирующие фак |
|||||||||||||||||||||||||||
имущества (например, |
рецепторы сте |
торы. Ключевую роль в их возникно |
||||||||||||||||||||||||||
роидных гормонов в раках молочной |
вении |
играют |
|
изменения |
|
регуляции |
||||||||||||||||||||||
железы и т. д.). |
|
|
|
|
|
|
|
|
клеточного |
цикла, |
апоптоза, |
|
мигра |
|||||||||||||||
И |
наконец, |
важнейшим |
призна |
ции и некоторых других базовых про |
||||||||||||||||||||||||
ком |
неопластических |
клеток |
являет |
цессов жизнедеятельности |
клетки. |
|
||||||||||||||||||||||
ся |
их |
генетическая |
|
нестабильность. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
Очевидно, |
что канцерогенез |
— |
мно |
2.2.1. |
Нарушения |
регуляции |
|
|||||||||||||||||||||
гоступенчатый |
|
процесс |
накопления |
|
||||||||||||||||||||||||
|
клеточного |
цикла |
|
|
|
|
|
|||||||||||||||||||||
мутаций и других генетических изме |
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
нений, |
приводящих |
к |
нарушениям |
Нарушения |
|
регуляции |
клеточного |
|||||||||||||||||||||
регуляции |
размножения |
и миграции |
цикла лежат в основе таких важней |
|||||||||||||||||||||||||
клеток, |
понижению |
|
их |
чувствитель |
ших свойств |
неопластической |
клетки, |
|||||||||||||||||||||
ности к различным ростсупрессирую- |
как самодостаточность в пролифера- |
|||||||||||||||||||||||||||
щим сигналам, ослаблению в них ин |
тивных |
сигналах |
|
(пониженная |
потреб |
|||||||||||||||||||||||
дукции |
апоптоза, |
|
блокированию |
ность во внешних сигналах для ини |
||||||||||||||||||||||||
дифференцировки |
|
и |
т. д. |
Вероят |
циации |
и |
поддержания |
пролифера |
||||||||||||||||||||
ность возникновения в одной клетке |
ции) и нечувствительность к ростсу- |
|||||||||||||||||||||||||||
нескольких |
генетических |
измене |
прессирующим |
сигналам. |
Кроме |
того, |
||||||||||||||||||||||
ний, |
|
создающих |
совокупность выше |
они в значительной степени опреде |
||||||||||||||||||||||||
указанных |
свойств, |
резко повышает |
ляют |
генетическую |
нестабильность |
и |
||||||||||||||||||||||
ся при нарушениях работы систем, |
нарушения |
дифференцировки |
клетки. |
|
||||||||||||||||||||||||
поддерживающих |
целостность |
гено |
В |
процессе |
|
подготовки |
клетки |
к |
||||||||||||||||||||
ма. |
Вследствие |
этого |
мутации, |
веду |
|
|||||||||||||||||||||||
делению и |
образования из |
нее двух |
||||||||||||||||||||||||||
щие |
к |
генетической |
|
нестабильности, |
||||||||||||||||||||||||
|
новых |
клеток |
наблюдается |
|
несколько |
|||||||||||||||||||||||
также |
|
являются |
неотъемлемым |
эта |
|
|||||||||||||||||||||||
|
фаз: |
G1 фаза, |
в которой |
идет |
подго |
|||||||||||||||||||||||
пом |
опухолевой |
прогрессии. Генети |
||||||||||||||||||||||||||
товка к синтезу Д Н К ; S фаза — пери |
||||||||||||||||||||||||||||
ческая |
нестабильность |
неопластиче |
||||||||||||||||||||||||||
од репликации Д Н К ; G2 фаза, в кото |
||||||||||||||||||||||||||||
ских |
|
клеток базируется |
на уменьше |
|||||||||||||||||||||||||
|
рой |
идет подготовка |
к митозу, |
|
и, на |
|||||||||||||||||||||||
нии |
точности |
воспроизведения |
гене |
|
||||||||||||||||||||||||
конец, |
собственно |
митоз |
— |
процесс |
||||||||||||||||||||||||
тического |
аппарата, |
нарушениях ме |
||||||||||||||||||||||||||
разделения |
клетки на две |
новые. Об |
||||||||||||||||||||||||||
ханизмов репарации Д Н К и |
измене |
|||||||||||||||||||||||||||
разовавшиеся |
дочерние |
клетки |
могут |
|||||||||||||||||||||||||
ниях |
|
регуляции |
клеточного |
цикла |
в |
|||||||||||||||||||||||
|
сразу войти |
в |
|
новый |
митотический |
|||||||||||||||||||||||
поврежденных |
клетках. Это вместе |
с |
|
|||||||||||||||||||||||||
цикл |
или временно |
выйти |
из |
|
него |
в |
||||||||||||||||||||||
уходом |
от |
апоптоза, |
позволяющим |
|
||||||||||||||||||||||||
GO фазу — стадию покоя. "Мотором" |
||||||||||||||||||||||||||||
генетически |
измененным |
клеткам |
||||||||||||||||||||||||||
клеточного |
цикла является |
активация |
||||||||||||||||||||||||||
выживать, |
делает |
популяции |
опухо |
|||||||||||||||||||||||||
последовательно сменяющих друг дру |
||||||||||||||||||||||||||||
левых |
клеток |
изменчивыми, |
создает |
|||||||||||||||||||||||||
га циклинзависимых |
киназ |
|
(схема |
|||||||||||||||||||||||||
основу для |
постоянного |
возникнове |
|
|||||||||||||||||||||||||
2.4). Каждая из циклинзависимых ки |
||||||||||||||||||||||||||||
ния и отбора все более и более злока |
||||||||||||||||||||||||||||
наз |
представляет собой |
холофермент- |
||||||||||||||||||||||||||
чественных |
вариантов. |
Таким |
обра |
|||||||||||||||||||||||||
ный |
комплекс, |
состоящий |
из |
собст |
||||||||||||||||||||||||
зом, |
генетическая нестабильность яв |
|||||||||||||||||||||||||||
венно |
каталитической |
субъединицы |
||||||||||||||||||||||||||
ляется двигателем |
неуклонной опухо |
|||||||||||||||||||||||||||
(cdk) |
и |
регуляторной субъединицы |
— |
|||||||||||||||||||||||||
левой |
прогрессии. |
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
циклина. |
Связывание |
с |
|
циклином |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
91
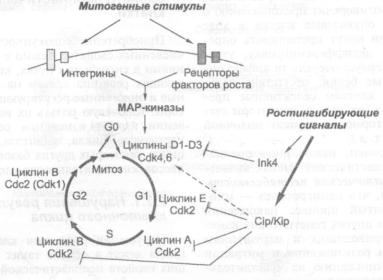
С х е м а 2.4. Общие принципы регуляции клеточного цикла в нор мальной клетке (объяснение в тексте)
увеличивает киназную активность cdk и, кроме того, определяет их локали зацию и субстратную специфичность. Уровень экспрессии каждого из циклинов и в меньшей степени cdk на правленно изменяется в определен ные фазы клеточного цикла. Так, вы ход клетки из стадии покоя и вход в фазу G1 определяется образованием комплексов циклинов D ( D l — D3) с cdk4 или cdk6 (в зависимости от типа клеток). Переход из G1 в S связан с образованием комплексов циклина Ε с cdk2, и т. д. Вход в митоз, например, обусловлен образованием комплексов cdk1 (другое и более распространен ное название — Cdc2) с циклином В.
Кроме связывания с циклинами, активность cdk регулируется измене ниями фосфорилирования их опреде ленных аминокислотных остатков (за такую регуляцию ответственны фосфатазы cdc25 и протеинкиназы САК, Weel) и связыванием с так называе мыми CKI — ингибиторами cdk. СК1 — группа белков, состоящая из
двух семейств. Представители семей ства Cip/Kip (p21WAFI/CIP1, р27КIР1 и р57КIР2) ингибируют различные ком
плексы cdk2, ответственные за вход и продвижение по фазе S. В меньшей степени они подавляют активность комплексов циклин B/cdc2, ответст венных за вход в митоз. С другой сто роны, они не ингибируют, а даже ак тивируют комплексы циклин D/
cdk4(6), оперирующие в ранней G1 фазе. Члены семейства Ink4 (pl6Ink4a, pl5ink4b, pl8lnk4c, pl9lnk4d) непосредст
венно взаимодействуют с cdk4(6). При этом, связывая cdk4(6), находящиеся в составе активных комплексов с цик лином D и белками Cip/Kip, они вы тесняют белки Cip/Kip, направляя их на связывание с cdk2. Поэтому повы шение активности белков 1nk4, в ча
стности опухолевого супрессора р161nk4а (см. раздел 2.2.3), вызывает как
прямое ингибирование активности комплексов циклин D/cdk4(6), так и непрямое блокирование комплексов циклин E/cdk2 и циклин A/cdk2.
Выход покоящейся клетки из фазы GO и вступление ее в митотический цикл инициируется различными внешними стимулами, в первую оче редь различными секретируемыми цитокинами, принадлежащими к группе
92
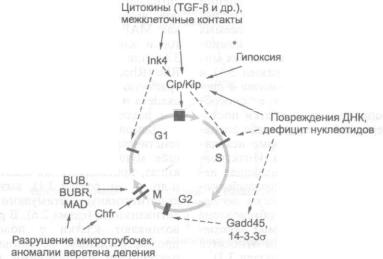
С х е м а 2.5. Активация чекпойнтов клеточного цикла (обозначе
ны черными прямоугольниками) при различных ростиигибирующих воздействиях
факторов роста. Кроме этого, для де ления большинства типов нормальных клеток необходимо также и взаимо действие специфических рецепторов клетки — интегринов — с определен ными белками внеклеточного матрикса (фибронектином, коллагеном и др.). Связывание рецепторов со своими лигандами — ростовыми фак торами и белками внеклеточного матрикса — индуцирует аутофосфорилирование внутриклеточных доменов ре цепторов и дальнейшее их взаимодей ствие со многими сигнальными бел ками (см. раздел 3.1). Следствием это го является стимуляция пересекаю щихся сигнальных путей и активация так называемых MAP-(Mitogen Acti vated Protein)киназных каскадов. Ко нечные продукты этих каскадов — се-
рин-треониновые киназы |
ERK1/2, |
J N K и р38 транспонируются |
из цито |
плазмы в ядро, где они фосфорилируют множество субстратов, что в конце концов и приводит к активации циклинзависимых киназ, инициирующих вход в фазу S.
Действие большинства ростингибирующих факторов (цитокинов типа
T G F - β , контактного торможения при установлении межклеточных контак тов, повреждений Д Н К и других внут риклеточных нарушений) основано на активации ингибиторов циклинзависимых киназ (CKI) семейств Ink4 и Cip/Kip (см. раздел 3.2), что приводит к остановке клеточного цикла в так называемых чекпойнтах (checkpoints — сверочные точки). В зависимости от типа ростингибирующего воздейст вия и молекул, вовлеченных в его рас познавание, наблюдается остановка клеточногр цикла в G l , S, G2 фазах или в митозе. При этом задержка в G2 связана как с активацией CKI семей ства Cip/Kip, так и с повышением ак тивности ряда других молекул, ингибирующих функцию комплексов циклин B/Cdc2 (см. раздел 3.2), а остановка в митозе — с изменениями ак тивности молекул, контролирующих конденсацию хромосом и разделение сестринских хроматид (схема 2.5).
Для опухолевых клеток характерны генетические изменения, вызываю щие, с одной стороны, перманентную стимуляцию сигнальных путей, акти вирующих cdk4(6) и cdk2, а с другой —
93
нарушения в путях передачи сигна лов, опосредующих активацию чекпойнтов в ответ на ростингибирующие сигналы. Первый тип изменений возникает в основном в результате ак тивирующих мутаций так называемых протоонкогенов — нормальных компо нентов путей передачи различных митогенных сигналов (см. раздел 3.1) и приводит к самодостаточности в пролиферативных сигналах, т. е. способ ности неопластической клетки посто янно генерировать внутри себя сигна лы к размножению, в норме исходя щие от внешних стимулов. Инактива ция чекпойнтов, обеспечивающая не чувствительность к ростингибирующим сигналам и генетическую неста бильность, чаще всего обусловлена дисфункцией так называемых опухоле вых супрессоров, к которым относятся
инекоторые из CKI (см. раздел 3.2).
Сизменениями регуляции клеточ ного цикла связаны и нарушения диф ференцировки неопластических клеток. Реализация большинства дифференцировочных программ требует выхода клетки из митотического цикла в ста дию покоя (GO). Перманентная сти муляция размножения клеток и не чувствительность к действию ростингибирующих цитокинов, являющихся одновременно индукторами диффе ренцировки (таких как TGF-p) неред ко приводят к блокированию или из вращению процессов дифференци ровки. (Некоторые другие механизмы нарушения дифференцировки неопла стических клеток рассмотрены в раз деле 3.2.1.2.)
2.2.2. Изменения морфологии и движения клеток
Верхние этажи сигнальных путей, активируемых связыванием рецепто ров ростовых факторов и интегринов со своими лигандами, ответственны за стимуляцию не только размножения, но и движения клеток. Поэтому мно гие из цитокинов являются одновре менно и митогенами, и мотогенами (например, E G F , H G F / S F , P D G F
и др.). Активация G-белков семейства Ras и фосфатидилинозит-3-киназы (PI3K), находящихся на пересечении сигнальных путей от многих рецепто ров, ведет к повышению активности как МАР-киназ — ключевых регуля торов клеточного цикла (см. раздел 3.1), так и малых ГТФ-аз семейства Rho (Rho, Rac, cdc42), играющих цен тральную роль в реорганизации цитоскелета и регуляции движения клеток (см. разделы 4.2, 4.3).
Для опухолевых клеток характерны генетические изменения (активирую щие мутации рецепторных тирозинкиназ, протоонкогенов семейства RAS и др. — см. раздел 3.1), вызывающие конститутивную стимуляцию такой сигнализации (схема 2.6). В результате возникают клетки с повышенным пролиферативным потенциалом, ха рактерными изменениями морфоло гии и увеличенной способностью к миграции, а следовательно, и к инвазивному росту и метастазированию. Клетка с такими изменениями, воз никшими, например, в результате ак тивации протоонкогенов семейства RAS, приобретает также и способность самостоятельно продуцировать и секретировать ряд митогенов/мотогенов, включая V E G F (Vascular Endotelial Growth Factor), стимулирующий раз множение и направленную миграцию окружающих эндотелиоцитов, т. е.
неоангиогенез.
2.2.3. Отсутствие репликативного старения (иммортализация)
Известно, что при культивирова нии in vitro человеческие фибробласты после 60—70 делений перестают размножаться и останавливаются, преимущественно в G1 фазе клеточ ного цикла. Очень редко, примерно в одной клетке из нескольких миллио нов, происходят спонтанные мутации, отменяющие остановку клеточного цикла. Однако потомки этой клетки делятся еще порядка 10 раз и снова останавливают свое размножение, а
94
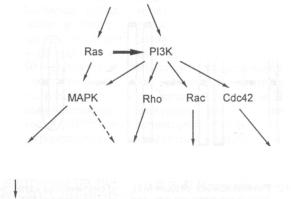
С х е м а 2.6. Верхние этажи сигнальных путей, активируемых ре
цепторами ростовых факторов. Контролируют размножение и движение клеток
Рецепторы митогенов/мотогенов
Циклинзависимые |
Стресс- |
Ламеллоподии |
Филоподии |
киназы |
фибриллы |
|
|
Пролиферация |
Изменения |
морфологии и |
движения |
затем постепенно гибнут. Первая ос тановка клеточного цикла получила название "ранний кризис", или стадия
M l , |
а вторая |
остановка и |
гибель кле |
ток |
стали |
определяться |
термином |
"кризис", или стадия М2, репликативного старения. В пользу того, что ос тановка и гибель клеток связана именно с числом предшествующих клеточных делений, а не просто с ас трономическим временем, проведен ным вне организма, в условиях in vitro свидетельствуют две группы фактов. Во-первых, в культурах фибробластов, полученных от эмбрионов или от мо лодых людей, кризис наступает позже, чем в культурах фибробластов, полу ченных от пожилых. Во-вторых, если до наступления стадии Ml фибробласты не пересаживать, они образуют плотную культуру и в результате кон тактного торможения остановятся в фазе G 1 . В этом состоянии они могут находиться довольно долго, по край ней мере 2—3 мес, а затем, после пе ресадки, снова начнут размножаться до тех пор, пока не пройдут свои 60— 80 делений. В это время их аналоги, не останавливавшие свое размноже ние, уже давно погибли. Таким обра
зом, астрономическое время пребыва ния в культуре может быть увеличено, а неизменным остается число клеточ ных делений до наступления кризиса. Если же какие-то клетки избегают кризиса (спонтанная частота такого события настолько низка, что ее даже трудно зарегистрировать, но опреде ленные манипуляции с геномом или экспрессия некоторых клеточных и вирусных онкогенов могут значитель но увеличить вероятность этого собы тия), то они могут размножаться уже бесконечно долго. Это определяется термином "иммортализация" клеток,
т.е. приобретение бессмертия.
Воснове счетно-ограничительного механизма, детерминирующего репликативное старение клеток, лежит про грессивное укорочение теломер (кон цевых участков хромосом) по мере де
ления клеток. Д Н К теломер, пред ставляющая собой более тысячи по второв гексануклеотида TTAGGG, в силу известной проблемы репликации концов линейной Д Н К синтезируется не полностью, и при каждом акте реп ликации, т. е. после каждого клеточ ного деления, теломеры укорачивают ся. Согласно теломерной гипотезе,
95
С х е м а 2.7. Теломерный механизм ренликативного старения
клеток человека
Неполная репликация концов линейной ДНК
Ранний кризис (стадия М1), |
«Генетическая катастрофа» |
остановка деления, как при |
(стадия М2); |
повреждениях ДНК |
слипание хромосом |
прогрессивное |
укорочение |
теломер |
основе |
лежит |
способность |
специфи |
||||||||||||
приводит к тому, что они достигают |
ческого |
фермента теломеразы |
дост |
|||||||||||||||
какой-то критической минимальной |
раивать |
недореплицированные |
тело- |
|||||||||||||||
длины, когда сенсорные системы на |
мерные повторы и поддерживать та |
|||||||||||||||||
чинают распознавать их как аномаль |
ким образом их постоянную длину. |
|||||||||||||||||
ные |
структуры |
Д Н К |
и |
индуцировать |
Теломераза |
состоит |
|
из |
|
нескольких |
||||||||
остановку |
клеточного |
цикла |
подобно |
субъединиц, включая РНК-матрицу и |
||||||||||||||
тому, |
как |
это |
происходит |
при |
Д Н К - |
TERT |
(Telomerase |
|
Reverse |
Tran |
||||||||
повреждающих |
воздействиях. |
|
Имен |
scriptase), представляющую |
собой об |
|||||||||||||
но это, как сейчас предполагается, и |
ратную |
транскриптазу, |
синтезирую |
|||||||||||||||
вызывает |
фазу |
|
Ml |
репликативного |
щую Д Н К |
повторов |
гексануклеотида |
|||||||||||
старения (ранний кризис). При нару |
T T A G G G с РНК-матрицы (подробнее |
|||||||||||||||||
шениях в сигнальных системах, детер |
см. раздел 3.5). Предполагается суще |
|||||||||||||||||
минирующих |
остановку |
клеточного |
ствование и других, так называемых |
|||||||||||||||
цикла, клетки с укороченными тело- |
АЬТ-(альтернативных) |
|
механизмов |
|||||||||||||||
мерами будут |
продолжать |
делиться, |
поддержания |
длины |
теломер, |
осно |
||||||||||||
пока теломеры практически не исчез |
ванных |
на |
гомологичной |
рекомбина |
||||||||||||||
нут и перестанут выполнять свои |
ции Д Н К |
теломер. |
Включение экс |
|||||||||||||||
функции, т. е. предотвращать реком |
прессии TERT, каталитической субъе |
|||||||||||||||||
бинации и слипание хромосом. Тогда |
диницы теломеразы, индуцируется из |
|||||||||||||||||
хромосомы |
потеряют |
|
свою |
|
целост |
менением |
экспрессии |
определенных |
||||||||||
ность и наступит так называемая ге |
онкогенов (см. раздел 3.1) или опухо |
|||||||||||||||||
нетическая |
катастрофа, |
или |
стадия |
левых супрессоров (см. раздел 2.3). |
||||||||||||||
М2 |
репликативного |
старения |
(схема |
Так, оно может быть вызвано актива |
||||||||||||||
2.7). |
|
|
|
|
|
|
|
|
|
цией онкогена Мус и инактивацией |
||||||||
Отсутствие |
в |
опухолевых |
|
клетках |
опухолевого |
|
супрессора |
р53. |
Кроме |
|||||||||
человека |
репликативного |
|
старения |
этого, существенный вклад в иммор- |
||||||||||||||
(иммортализация) |
связано |
с |
включе |
тализацию |
неопластических |
клеток |
||||||||||||
нием |
специального механизма. В его |
вносят |
нарушения |
работы |
охранных |
|||||||||||||
96
механизмов, осуществляющих оста новку клеточного цикла при наруше ния структуры Д Н К , в частности при исчезновении теломерных повторов (см. раздел 3.2).
При обычных условиях культиви ровании in vitro в некоторых типах клеток человека, например в эпителиоцитах, наблюдается остановка кле точного цикла через 10—15 делений — так называемая фаза МО репликатив ного старения. Такая остановка не связана, по-видимому, с изменением
длины теломер, а обусловливается ак тивацией CKIs, в частности pl6lnk4a, в
ответ на какие-то внутриклеточные изменения, вызываемые неадекватно стью условий in vitro. Так фаза МО в кератиноцитах или эпителиоцитах мо лочной железы может быть отменена при культивировании их на подложке фибробластов в среде без сыворотки, но с достаточным содержанием набо ра определенных факторов роста. Сходная ситуация наблюдается в культурах фибробластов и других кле ток грызунов, которые входят в кри зис через 10—15 делений in vitro, не смотря на очень большую длину теломер и высокую активность теломеразы в подавляющем большинстве клеток. Как и в случае эпителиальных клеток человека, кризис в клетках грызунов может быть предотвращен подбором адекватных условий культивирования (определенный для каждого типа кле ток внеклеточный матрикс и набор цитокинов), в связи с чем он получил название "культуральный шок" (cul ture shock). Таким образом, совсем не давно выяснилось, что у клеток гры зунов в отличие от клеток человека нет механизма подсчета числа деле ний (т. е. они как бы изначально иммортализованы вследствие конститу тивной активности теломеразы, под держивающей довольно большую дли ну теломер), а их репликативное ста рение представляет собой артефакт, связанный с неадекватностью условий in vitro. Описанные ранее события, вызывающие иммортализацию фиб робластов грызунов (инактивация
опухолевых супрессоров р53, pARF — см. раздел 3.2) или отмену фазы МО в
эпителиоцитах человека (инактивация опухолевых супрессоров pl6lnk4a, pRb —
см. раздел 3.2), предотвращают акти вацию чекпойнтов клеточного цикла в ответ на внутриклеточные изменения, накапливающиеся при неправильном культивировании клеток. Кроме того, они могут отменять индукцию терми нальной дифференцировки фактора ми сыворотки крупного рогатого ско та, которая является причиной артефактного репликативного старения некоторых типов клеток, например крысиных олигодендроцитов и шванновских клеток.
2.2.4. Изменения регуляции апоптоза
Апоптоз вызывается различными сигналами, как физиологическими — экспрессией специальных киллерных цитокинов, изменениями гормональ ного статуса (цикличное ремоделирование эндометрия матки, возрастная инволюция тимуса и др.), так и нефи зиологическими — внутриклеточными повреждениями или неблагоприятны ми условиями: нехваткой факторов роста, повреждениями Д Н К , гипокси ей и т. д. В регуляции апоптоза выде ляют два основных этапа: фазу индук ции (принятия решения) и фазу экзе куции (исполнения приговора). По следняя осуществляется путем актива ции каспаз — семейства цистеиновых протеиназ, расщепляющих свои суб страты по остаткам аспартатовой ки слоты. Расщепление каспазами 3, 6, 7 (так называемые эффекторные, или казнящие каспазы) ряда ключевых субстратов, в частности ингибиторов нуклеаз, ламинов — ядерных цитоскелетных белков и т. д., приводит к фрагментации Д Н К и деструкции клетки. Каспазы присутствуют в цито плазме в виде проэнзимов и активи руются до полностью функциональ ных протеаз путем расщепления проэнзима на большую и малую субъеди ницы и дальнейшего отщепления от
7-7908 Д. Г. Заридзе |
97 |
|

Цитохром С  Каспаза 9
Каспаза 9
Ключевые субстраты
Апоптоз
них N-концевых доменов. Затем субъ единицы собираются в активные олигомеры. Расщепление прокаспаз могут осуществлять различные протеазы, в том числе и другие каспазы.
Существует два принципиально разных сигнальных пути, приводящих к активации эффекторных каспаз 3, 6, 7 (схема 2.8). Один из них иницииру
ется |
связыванием |
специфических |
|
киллерных |
лигандов |
(Fas-лиганд, |
|
T N F - a и др.) |
со своими рецепторами, |
||
так |
называемыми рецепторами смер |
||
ти, что вызывает рекрутирование к ним адаптерных белков и прокаспаз, в частности прокаспазы 8 (см. раздел 3.4). Агрегация молекул прокаспазы 8 достаточна, чтобы инициировать их аутопроцессирование (расщепление) и образование активных форм каспазы 8, которая в свою очередь процессирует до активных форм эффекторные каспазы.
При альтернативном пути индук ции апоптоза ключевую роль играют митохондрии, поэтому его называют митохондриальным путем. Он ини циируется главным образом различ ными повреждающими воздействия ми, вызывающими увеличение прони цаемости митохондриальной мембра ны и выход в цитоплазму ряда митохондриальных белков, в частности ци-
тохрома С, который связы вается с белком APAF-1 и стимулирует образование его оли гомеров. Это в свою очередь вызывает рекрути рование на образовавший ся комплекс молекул про каспазы 9, их агрегацию, аутопроцессирование и формирование активного комплекса каспазы 9. На следующем этапе происхо дит рекрутирование на этот комплекс молекул прокас пазы 3 и их процессирование до активных форм, ко торые расщепляют ключе вые мишени и вызывают апоптоз. Следует заметить, что повреждения и стрессы
приводят к выходу из митохондрий не только цитохрома С, но и ряда других молекул, в частности протеазы AIF (Apoptosis Inducing Factor). A1F также стимулирует апоптоз, вызывая расще пление ряда ядерных белков каспазонезависимым путем. Таким образом, митохондриальный путь индукции апоптоза включает несколько незави симых механизмов (схема 2.9). При этом существует взаиморегуляция сиг нальных путей, активируемая рецеп торами смерти и повреждающими воздействиями (см. схему 2.8). Так, активация каспазы 8, вызванная акти вацией рецепторов смерти, не только напрямую активирует эффекторные каспазы, но и индуцирует увеличение проницаемости митохондриальной мембраны и активацию регуляторной каспазы 9. В то же время при повреж дениях и стрессах может наблюдаться повышение экспрессии рецепторов смерти — Fas и Killer/DR5. Таким об разом, проапоптотические сигналы амплифицируются, что позволяет на дежнее достигать необходимого эф фекта.
Ключевую роль в регуляции про ницаемости митохондриальной мем браны для цитохрома С и AIF играют белки семейства bcl-2, подразделяю щиеся на несколько подсемейств и
98
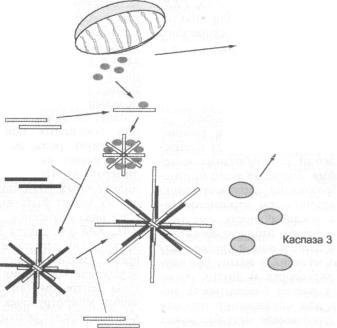
С х е м а 2.9. Механизмы митохондриального пути индукции
апоптоза
Каспазонезависимый
апоптоз
Цитохром С |
AIF |
APAF-1
Каспазозависимый
апоптоз
Прокаспаза 9
Прокаспаза 3
обладающие либо проапоптотическими, либо антиапоптотическими актив ностями. Предполагается, что антиапоптотические белки (bcl-2, bcl-X, и др.), локализуясь в мембранах мито хондрий, закрывают каналы, через ко торые осуществляется выброс цито хрома С и AIF. Проапоптотические молекулы (Вах, Bad и др.) при апоптогенных сигналах перемещаются из цитоплазмы в митохондриальные мембраны, где, взаимодействуя с ин тегральным белком наружной мито хондриальной мембраны VDAC, сти мулируют открытие канала, через ко торый, в частности, секретируется ци тохром С. Кроме того, белки подсе мейства Вах образуют гетеромерные комплексы с белками bcl-2, bcl-x, что, возможно, открывает закрытые до этого каналы.
Для опухолевых клеток характерны генетические изменения, ведущие к ослаблению обоих путей индукции
апоптоза. Так, в них закономерно об наруживаются:
•потеря экспрессии на поверхно
сти клетки рецептора смерти Fas;
•нарушения проведения апоптогенного сигнала к митохондриям (например, при инактивации опухолевых супрессоров р53 и PTEN — см. раздел 3.2);
•ингибирование проницаемости
митохондриальной |
мембраны |
|
для |
цитохрома С и A1F вследст |
|
вие |
изменений экспрессии бел |
|
ков семейства bcl-2 (см. разделы 2.1, 3.1);
• блокирование активации эсрфекторных каспаз (например, при потере экспрессии белка APAF- 1 в результате метилирования его гена — см. раздел 3.6);
•* резкое уменьшение времени жизни каспаз ввиду их связыва-
7* |
99 |
|
ния с белками ΙΑΡ (Inhibitors of |
Совокупность |
перечисленных |
на |
||||||||||||||||
|
Apoptosis), |
экспрессия |
которых |
рушений |
обеспечивает |
|
повышенную |
|||||||||||||
|
повышается в результате актива |
частоту возникновения |
различных ге |
|||||||||||||||||
|
ции протоонкогенов |
RAS, |
|
РКВ/ |
нетических изменений и их закрепле |
|||||||||||||||
|
Akt (см. раздел 3.1) или инакти |
ние в ряду клеточных поколений. |
||||||||||||||||||
|
вации |
опухолевого |
супрессора |
Понижение точности |
|
репликации |
||||||||||||||
|
PTEN |
(см. раздел 3.2). |
|
|
|
Д Н К |
в неопластических |
|
клетках |
свя |
||||||||||
|
|
|
|
|
|
|
|
|
|
зано с повышением синтеза и актив |
||||||||||
2.2.5. Генетическая |
|
|
ности |
в них так |
называемых низко |
|||||||||||||||
|
|
точных полимераз, в частности ДНК- |
||||||||||||||||||
нестабильность |
|
|
|
|
||||||||||||||||
|
|
|
|
полимеразы-β, которая в норме ис |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||
Генетическая |
нестабильность |
— |
пользуется лишь для быстрой репара |
|||||||||||||||||
это |
увеличение |
вероятности |
возник |
ции |
массивных |
повреждений |
ДНК |
|||||||||||||
новения и закрепления в ряду клеточ |
(основную |
роль |
в |
воспроизводстве |
||||||||||||||||
ных |
поколений |
разнообразных |
изме |
Д Н К |
играет высокоточная ДНК-по- |
|||||||||||||||
лимераза-δ). Повышение |
содержания |
|||||||||||||||||||
нений генома. Значение этого призна |
||||||||||||||||||||
низкоточных ДНК-полимераз-β, |
-ε и |
|||||||||||||||||||
ка для образования |
злокачественной |
|||||||||||||||||||
других может быть вызвано |
экспрес |
|||||||||||||||||||
опухоли связано с тем, что именно ге |
||||||||||||||||||||
сией |
ряда онкогенов, |
например |
ВСR/ |
|||||||||||||||||
нетическая |
нестабильность |
|
вместе с |
|||||||||||||||||
|
ABL, RAS (см. раздел |
3.1). Нарушения |
||||||||||||||||||
постоянно идущим отбором обеспечи |
||||||||||||||||||||
правильной сегрегации |
хромосом во |
|||||||||||||||||||
вают накопление в одной клетке сразу |
||||||||||||||||||||
время |
митоза |
могут |
|
|
происходить |
|||||||||||||||
нескольких мутаций в онкогенах, опу |
|
|
||||||||||||||||||
вследствие |
изменения |
числа |
и струк |
|||||||||||||||||
холевых супрессорах |
и |
других |
генах, |
|||||||||||||||||
туры |
центросом |
или |
центров |
органи |
||||||||||||||||
придающих |
клетке |
совокупность |
не |
|||||||||||||||||
зации |
микротрубочек: |
в |
опухолевых |
|||||||||||||||||
обходимых |
для |
образования |
опухоли |
|||||||||||||||||
клетках нередко обнаруживается боль |
||||||||||||||||||||
свойств. Генетическая нестабильность |
||||||||||||||||||||
ше двух центросом, что ведет к много- |
||||||||||||||||||||
популяций |
опухолевых |
клеток |
скла |
|||||||||||||||||
полярным |
митозам и |
возникновению |
||||||||||||||||||
дывается из |
4 основных |
типов |
нару |
|||||||||||||||||
анеуплоидных вариантов с неправиль |
||||||||||||||||||||
шений: |
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
ным числом хромосом. К увеличению |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||
• |
уменьшения точности |
воспроиз |
числа центросом в клетке приводит |
|||||||||||||||||
|
ведения генетической |
информа |
активация онкогена RAS и инактива |
|||||||||||||||||
|
ции, а именно — понижения точ |
ция опухолевых супрессоров р53, АРС |
||||||||||||||||||
|
ности репликации Д Н К и сегре |
или BRCA1 (см. раздел 3.2). Осталь |
||||||||||||||||||
|
гации хромосом во время митоза; |
ные компоненты |
генетической неста |
|||||||||||||||||
•нарушений в системах репара бильности неопластических клеток —
ции |
повреждений Д Н К или |
|
ошибок, возникших при ее реп |
||
ликации; |
|
|
• ослабления функции |
чекпойн- |
|
тов |
клеточного цикла, |
активи |
руемых в ответ на повреждения |
||
структуры Д Н К или веретена де |
||
ления в митотической клетке, в |
||
результате чего клетка, |
несмотря |
|
на разрывы Д Н К или изменения числа хромосом, продолжает де литься и умножать число ано мальных потомков;
нарушения работы систем репарации Д Н К , инактивация чекпойнтов кле точного цикла и ослабление индукции апоптоза — рассмотрены в других раз делах (см. разделы 3.2.11, 2.2.1 и 2.2.4).
В последние годы выяснилось, что повышенная изменчивость популяций опухолевых клеток связана не только с резким учащением появления ис тинных генетических изменений (ген ных мутаций, рекомбинаций, анеуплоидии и др.), но и со значительным
•ослабления индукции апоптоза, увеличением вероятности возникнове
вследствие чего делящиеся клет ки с генетическими нарушения ми не погибают, а выживают.
ния в неопластических клетках так называемых эпигенетических измене ний. В основе таких изменений лежит
100
ремоделирование структуры хромати на, обусловленное изменениями ме тилирования цитозинов ДНК и/или ацетилирования гистонов. В результа те происходит подавление экспрессии одних генов и/или повышение экс прессии других, причем из-за харак терных для опухолевых клеток нару шений процессов метилирования Д Н К одновременно может изменяться транскрипция нескольких сотен генов, (подробнее см. раздел 3.6).
* * *
Итак, канцерогенез — это многосту пенчатый процесс накопления мутаций и других генетических изменений, при водящих к нарушениям регуляции кле точного цикла, апоптоза, дифференци ровки, движения и морфогенетических реакций клетки, а также контроля це лостности генома. Все это в свою оче редь обеспечивает приобретение клет кой и ее потомками ряда свойств, та ких как самодостаточность в пролиферативных сигналах, повышение ми грационной способности, нечувстви тельность к ростингибирующим воз действиям, отсутствие репликативно го старения, увеличение жизнеспособ ности при неблагоприятных условиях окружающей среды или внутрикле точных повреждениях, генетическая нестабильность и т. д., которые детер минируют неопластическую транс формацию и дальнейшую опухолевую прогрессию. Ключевую роль в воз никновении указанных свойств не опластической клетки играют наруше ния функции протоонкогенов (см. раздел 3.1) и опухолевых супрессоров (см. раздел 3.2). Исследования по следних лет позволили идентифици ровать сигнальные пути, контролируе мые большинством из этих генов. Вы яснилось, что многие из них регули руют активность одних и тех же путей на разных этажах передачи сигналов, что некоторые из таких сигнальных путей одновременно вовлечены в ре гуляцию нескольких важнейших фи зиологических процессов. Например,
активация протоонкогенов семейства RAS и регулируемых им путей переда чи сигналов не только стимулирует клеточную пролиферацию (см. раздел 3.1), но и вызывает изменения формы и подвижности клеток (см. раздел 6.3), а в некоторых клеточных контек стах — также и подавление апоптоза. В то же время продукты некоторых из опухолевых супрессоров и протоонко генов являются местами пересечения различных сигнальных путей. Так, р53, активируясь в ответ на самые разные повреждающие и стрессовые воздействия, взаимодействует с раз личными мишенями и контролирует апоптоз, продвижение по клеточному циклу, стабильность генома, локомо торные реакции и дифференцировку клеток (см. раздел 3.2.2). Отсюда ста новится понятной частое фиксирова ние изменений генов р53 и RAS в са мых разных новообразованиях: их му тации позволяют одномоментно при дать клетке сразу несколько свойств, определяющих опухолевую прогрес сию.
Однако для ряда новообразований, и в первую очередь для лейкозов, ха рактерны генетические изменения, встречающиеся только при данном за болевании. К ним относятся прежде всего хромосомные транслокации, пе ремещающие протоонкогены и/или опухолевые супрессоры в другое место генома (см. раздел 3.1). Специфич ность таких изменений может быть обусловлена рядом причин: а) увели ченной вероятностью некоторых гене тических перестроек в определенных типах клеток (например, соединение протоонкогена MYC с генами имму ноглобулинов является закономерной ошибкой перестройки генов иммуног лобулинов в ходе дифференцировки В-лимфоцитов — см. раздел 3.1); б) тканеспецифичным характером экс прессии или действия определенных онкогенов/опухолевых супрессоров; в) необходимостью приобретения разны ми типами клеток специфических на боров биологических свойств, обеспе чивающих их малигнизацию. Вероят-
101
но, исследование тканеспецифических механизмов канцерогенеза в бли жайшее время станет одной из наибо лее бурно развивающихся областей онкологии.
Рекомендуемая литература
Копнин Б. П. Мишени действия онкогенов и опухолевых супрессоров: ключ к по
ниманию базовых |
механизмов |
канце |
|||||
рогенеза (обзор) // Биохимия.'— 2000. — |
|||||||
Т. 65. |
- |
С. 5 - 3 3 . |
|
|
|
|
|
Blume-Jensen P., Hunter |
Т. |
Oncogenic |
kinase |
||||
signaling // Nature. — 2001. — Vol. 411. — |
|||||||
P. 355365 . |
|
|
|
|
|
||
Datta S. R., |
Brunet A., |
Greenberg |
M. |
E. |
Cellu |
||
lar survival: a play in three Akts // Genes |
|||||||
Dev. |
- |
1999. - |
Vol. |
13. |
- |
P. |
2 9 0 5 - |
2927. |
|
|
|
|
|
|
|
DePinho R. A. The age of cancer // Nature. —
2000. - Vol. 408. - P. 248-254. |
|
|
||||||
Enver |
Т., |
Greaves |
M. |
Loops, |
lineage, |
and |
||
leukemia |
// Cell. - |
1998. |
- Vol. 94. |
- |
||||
P. |
9 - 1 2 . |
|
|
|
|
|
|
|
Evan |
G. |
I, |
Vousden |
К. H. Proliferation, |
cell |
|||
cycle and apoptosis in cancer // Nature. — |
||||||||
2001. - Vol. 411. - P. 342-348. |
|
|
||||||
Gray J. W., Collins C. Genome |
changes and |
|||||||
gene expression in human solid tumors // |
||||||||
Carcinogenesis. — 2000. — Vol. 21. — |
||||||||
P. 443 - 452 . |
|
|
|
|
|
|||
Green |
D. R. Apoptotic pathways: paper wraps |
|||||||
stone blunts scissors // Cell. |
— 2000. |
— |
||||||
Vol. 102. |
- P. |
1-4. |
|
|
|
|
||
Green D. R. Apoptotic pathways: the roads to
ruin |
// |
Cell. |
- |
1998. |
- |
Vol. |
94. |
- |
|
P. 69.5-698. |
|
|
|
|
|
|
|
||
Hanahan |
D., |
Weinberg |
R. A. The hallmarks of |
||||||
cancer // Cell. — 2000. — Vol. 100. — |
|||||||||
P. 57 - 70 . |
|
|
|
|
|
|
|
||
Heldin C.-H. Signal |
transduction: |
multiple |
|||||||
pathways, |
multiple |
options |
for |
therapy |
// |
||||
Stem |
Cells. |
- |
2001. |
- |
Vol. |
19. |
- |
||
P. 295 - 303 . |
|
|
|
|
|
|
|
||
Hengartner M. |
O. |
The |
biochemistry |
of |
apop |
||||
tosis // Nature. — 2000. |
- |
Vol. |
407. |
- |
|||||
P. 770-776. |
|
|
|
|
|
|
|
||
Lengauer |
C., |
Kinzler K. |
W., Vogelstein B. Ge |
||||||
netic instabilities in human cancers // Na ture. - 1998. - Vol. 396. - P. 643-649.
McClatchey A. I. Modeling metastasis in the mouse // Oncogene. — 1999. — Vol. 18. — P. 5334-5339.
Ponder B. A. J. Cancer genetics // Nature. — 2001. - Vol. 411. - P. 336 - 341 .
Roovers K., Assoian R. K. Integrating the MAP
kinase |
signal |
into |
the |
Gl phase cell cycle |
|||
machinery // BioEssays. — 2000. — |
|||||||
Vol. 22. - P. 818-826. |
|
|
|||||
Saaristo A., |
|
Karpanen |
Т., |
Alitalo |
K. |
Mecha |
|
nisms of angiogenesis and their use in the |
|||||||
inhibition of tumor growth and metasta |
|||||||
sis // Oncogene. — 2000. — Vol. 19. - |
|||||||
P. 6122-6129. |
|
|
|
|
|||
Schmitt C. A., Lowe S. |
W. Apoptosis and ther |
||||||
apy // |
J. |
Pathol.. |
- |
1999. - |
Vol. |
187. - |
|
P. 127-137. |
|
|
|
|
|
||
Schwartz M. A., Baron |
V. |
Interactions |
between |
||||
mitogenic stimuli, or, a thousand and one |
|||||||
connections // Curr. Opin. Cell |
Biol. — |
||||||
1999. - |
Vol. |
11. |
- P. 197-202. |
|
|||
Shay J. W., |
Wright D. E. |
When |
do telomeres |
||||
matter? // Science. — 2001. — Vol. 291. - P. 839-840.
Sherr C. J. Cancer cell cycles // Science. —
1996. - |
Vol. 274. - P. 1672-1677. |
|
||||||||
Sherr |
C. J. |
The |
Pezcoller |
Lecture: |
Cancer |
|||||
Cell Cycles Revisited // Cancer Res.. — |
||||||||||
2000. - Vol. 60. - P. 3689-3695. |
|
|
||||||||
Sherr |
C. J., |
DePinho |
R. A. |
Cellular |
Senes |
|||||
cence: Mitotic Clock or Culture Shock? // |
||||||||||
Cell. - 2000. |
- |
Vol. |
102. - P. 407-410. |
|||||||
Sherr |
C. J., |
Roberts |
|
J. |
M. |
C D K |
inhibitors: |
|||
positive and negative regulators of Gl - |
||||||||||
phase progression // Gen. Dev. — 1999. — |
||||||||||
Vol. 13. - P. 1501-1512. |
|
|
|
|||||||
Stambolic V., |
Mak |
T. |
W., |
Woodgett J. R. Mod |
||||||
ulation of cellular apoptotic potential: con |
||||||||||
tributions to oncogenesis // Oncogene. — |
||||||||||
1999. - |
Vol. 18. - P. 6094-6103. |
|
|
|||||||
The |
Genetic |
Basis of Human Cancer / Eds B. |
||||||||
Vogelstein, K. W. Kinzler. — New |
York: |
|||||||||
McGraw Hill, |
1998. |
|
|
|
|
|
||||
Zhu |
L., Skoultchi A. |
I. |
Coordinating |
cell |
pro |
|||||
liferation |
and |
|
differentiation |
// |
|
Curr. |
||||
Opin. Genet. Dev. — 2001. — Vol. 11. -
P. 91 - 97 .
Г л а в а 3 МОЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЕ ИЗМЕНЕНИЯ
ВЗЛОКАЧЕСТВЕННЫХ КЛЕТКАХ
А.Г. Татосян
3.1. Онкогены |
|
|
|
|
|
|
|
ции онкогенов. Другие генетические |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
факторы |
канцерогенеза |
представлены |
|||||||||
В настоящее время можно считать |
в разделах 2; 3.2; 12.2. |
|
|
|
|
|
|||||||||||||||||||
доказанным, что возникновение, тем |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
пы роста опухоли и ее прогрессия оп |
3.1.1. |
Как |
|
|
|
|
|
|
|
||||||||||||||||
ределяются |
изменениями |
структурных |
|
|
|
|
|
|
|
||||||||||||||||
идентифицируются |
|
|
|
||||||||||||||||||||||
компонентов |
|
генома |
клетки. Эти из |
|
|
|
|||||||||||||||||||
|
онкогены? |
|
|
|
|
|
|
||||||||||||||||||
менения могут являться |
аберрациями, |
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
закрепленными в геноме, т. е. насле |
Активный |
поиск |
|
генетических |
|||||||||||||||||||||
дуемыми в поколениях, либо появ |
компонентов, |
ответственных за злока |
|||||||||||||||||||||||
ляться de novo в клетке и стимулиро |
чественную |
трансформацию |
клеток, |
||||||||||||||||||||||
вать развитие |
опухолевого |
процесса. |
был |
обоснован рядом |
основополагаю |
||||||||||||||||||||
К |
подобным |
генетическим |
|
струк |
щих теоретических положений, сфор |
||||||||||||||||||||
турным |
компонентам |
клетки, |
вовле |
мулированных в 50—60-е годы. К ним |
|||||||||||||||||||||
ченным |
в |
канцерогенез |
(онкогенным |
относятся |
|
вирусгенетическая |
теория |
||||||||||||||||||
детерминантам), можно отнести: |
Зильбера, гипотеза Хюбнера и Тодаро |
||||||||||||||||||||||||
а) |
протоонкогены |
— |
нормальные |
о вирогене |
и |
онкогене, |
провирусная |
||||||||||||||||||
гипотеза |
Темина, |
гипотеза |
о |
клеточ |
|||||||||||||||||||||
клеточные |
гены, |
участвующие |
в |
клю |
|||||||||||||||||||||
ном происхождении вирусных онкоге |
|||||||||||||||||||||||||
чевых |
|
процессах |
|
жизнедеятельности |
|||||||||||||||||||||
|
|
нов и др. |
|
|
|
|
|
|
|
|
|
||||||||||||||
клетки: регуляции транскрипции, рос |
|
|
|
|
|
|
|
|
|
||||||||||||||||
Онкогены |
как |
специфический |
ге |
||||||||||||||||||||||
та, клеточного |
цикла, |
передаче сигна |
|||||||||||||||||||||||
нетический |
|
материал, |
кодирующие |
||||||||||||||||||||||
ла и т. д. В |
случае |
структурных |
изме |
|
|||||||||||||||||||||
информацию об определенном белко |
|||||||||||||||||||||||||
нений |
|
или |
при |
повышении |
уровня |
||||||||||||||||||||
|
вом |
продукте, |
впервые были |
иденти |
|||||||||||||||||||||
экспрессии |
|
этих |
|
генов |
нарушается |
||||||||||||||||||||
|
|
фицированы в составе ретровирусов. |
|||||||||||||||||||||||
контроль |
нормального |
|
клеточного |
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
роста и дифференцировки, что приво |
Геном |
типичного |
неонкогенного |
||||||||||||||||||||||
дит к трансформации клетки. Такие |
ретровируса |
представляет собой моле |
|||||||||||||||||||||||
активированные |
протоонкогены |
при |
кулу односпиральной Р Н К , содержа |
||||||||||||||||||||||
нято называть онкогенами; |
|
|
|
щей три гена: ген gag, кодирующий |
|||||||||||||||||||||
б) |
гены-супрессоры |
|
(антионкоге |
структурные |
белки |
вирусной |
частицы; |
||||||||||||||||||
|
ген |
env, кодирующий |
белки |
оболочки |
|||||||||||||||||||||
ны) — |
гены, |
кодирующие |
ключевые |
||||||||||||||||||||||
вириона, и ген pol, |
несущий информа |
||||||||||||||||||||||||
регуляторные |
|
белки, |
потеря |
которых |
|||||||||||||||||||||
|
цию |
о ревертазе. |
Ревертаза |
|
является |
||||||||||||||||||||
влечет |
за собой нарушения |
контроля |
|
||||||||||||||||||||||
ферментом, |
имеющим |
комплекс раз |
|||||||||||||||||||||||
пролиферации; |
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
личных свойств, которые обеспечива |
|||||||||||||||||
в) |
гены-модуляторы |
— |
|
гены, |
спо |
||||||||||||||||||||
|
ют |
превращение |
вирусной |
|
Р Н К |
в |
|||||||||||||||||||
собствующие |
распространению |
опухо |
|
||||||||||||||||||||||
форму Д Н К |
провируса. |
На |
концах |
||||||||||||||||||||||
ли в организме, но |
не отвечающие за |
||||||||||||||||||||||||
провируса |
имеются |
повторяющиеся |
|||||||||||||||||||||||
злокачественную |
|
|
трансформацию |
||||||||||||||||||||||
|
|
последовательности — |
длинный кон |
||||||||||||||||||||||
клетки |
непосредственно. |
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
цевой повтор (LTR), размер которого |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
В данной главе будут рассмотрены |
варьирует от 300 до 1300 оснований у |
||||||||||||||||||||||||
современные |
|
представления |
о |
|
строе |
различных ретровирусов. Эта |
структу |
||||||||||||||||||
нии, |
свойствах и |
механизмах |
актива |
ра определяет |
ряд |
важнейших |
свойств |
||||||||||||||||||
103
Т а б л и ц а |
3.1. Онкогены, трансдуцирован- |
ретровирусного |
генома: интеграцию в |
|||||||||||||||
ные ретровирусами |
|
|
|
различные точки Д Н К клетки, регуля |
||||||||||||||
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
цию экспрессии вирусных генов, реп |
||||||||||||
Ген/локус |
|
Вирусы |
Ассоциирован |
|||||||||||||||
|
ликацию вируса как относительно са |
|||||||||||||||||
|
ные неоплазии |
|||||||||||||||||
|
|
|
||||||||||||||||
|
|
|
|
|
|
мостоятельного, генетически |
незави |
|||||||||||
Abl |
|
Ab-MuLV/ |
Т-клеточная |
|||||||||||||||
|
симого |
элемента. |
|
|
|
|
|
|
||||||||||
|
|
HZ2-FeSV |
лимфома/сар- |
Таким |
образом, |
|
ретровирусы |
со |
||||||||||
|
|
|
кома |
|
|
держат |
всю |
генетическую |
информа |
|||||||||
Akt |
|
AKT8 |
Тимома |
|
|
цию, |
необходимую для |
их |
полноцен |
|||||||||
Сbl |
|
Cas NS-1 |
В-клеточные |
|||||||||||||||
|
ного |
существования |
и |
репродукции в |
||||||||||||||
|
|
|
лимфомы |
|
|
|||||||||||||
|
|
|
|
|
зараженных |
клетках. |
|
|
|
|
|
|||||||
Суl-1 |
|
MuLV |
Лимфомы |
|
|
|
|
|
|
|
||||||||
|
|
|
Геном |
ретровируса |
имеет |
следую |
||||||||||||
Сrk |
|
ASV CT10 |
Саркома |
|
|
|||||||||||||
|
|
|
щую общую структуру: |
|
|
|
|
|||||||||||
ErbA/ErbB |
|
AEV-ES4 |
Эритролейкоз |
|
|
|
|
|||||||||||
|
LTR-gag-pol-env-UTR |
|
|
|
|
|
||||||||||||
ErbB |
|
ALV/ |
и |
|
|
|
|
|
|
|
||||||||
|
|
|
Ряд |
уникальных |
свойств ретрови |
|||||||||||||
|
|
RPL25/ |
|
|
|
|||||||||||||
|
|
|
|
|
русов |
(способность |
существования в |
|||||||||||
|
|
RPL28 |
|
|
|
|||||||||||||
|
|
|
|
|
форме Р Н К и Д Н К , интеграция в ге |
|||||||||||||
Ets |
|
AEV-E26 |
и |
|
|
|||||||||||||
|
|
|
ном клетки, наличие ревертазы, высо |
|||||||||||||||
Fgr |
|
GR-FeSV |
Саркома |
|
|
|||||||||||||
|
|
|
кая |
частота |
рекомбинаций |
на |
геном |
|||||||||||
Fms |
|
SM-FeSV и |
|
|
|
|||||||||||||
|
|
|
|
ном |
уровне) |
сделал |
их |
естественными |
||||||||||
|
|
HZ5-FeSV |
|
|
|
|||||||||||||
|
|
|
|
|
потенциальными векторами для пере |
|||||||||||||
Fos |
|
FBJ- и |
" |
|
|
|||||||||||||
|
|
|
носа генетической информации (в ча |
|||||||||||||||
|
|
FBR-MuSV |
|
|
|
|||||||||||||
|
|
|
|
|
стности, |
клеточных |
генов). |
Именно |
||||||||||
Fps/Fes |
|
FSV |
" |
|
|
|||||||||||||
|
|
|
такими |
носителями |
клеточных генов |
|||||||||||||
Jun |
|
ASV 17 |
Т-клеточная |
|||||||||||||||
|
являются онкогенные ретровирусы. В |
|||||||||||||||||
|
|
|
лимфома |
|
|
|||||||||||||
|
|
|
|
|
настоящее время известно |
более двух |
||||||||||||
Kit |
|
HZ4-FeSV |
Саркома |
|
|
|||||||||||||
|
|
|
десятков онкогенов, выявленных в ге |
|||||||||||||||
Maf |
|
AS42 |
|
|
|
|||||||||||||
|
|
|
|
номе |
различных ретровирусов (табл. |
|||||||||||||
Mos |
|
Mo-MuSV |
В-клеточная |
|||||||||||||||
|
3.1). |
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
лимфома/сар- |
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
Принципиально возможны два ти |
|||||||||||||||
|
|
|
кома |
|
|
|||||||||||||
|
|
|
|
|
па |
включения |
инородной |
генетиче |
||||||||||
Mpl |
|
MyLV |
Эритролейкоз |
|||||||||||||||
Myb |
|
MuLV, |
В-клеточная |
ской информации в состав вирусного |
||||||||||||||
|
|
ALV |
лимфома, |
мие- |
генома: |
присоединение дополнитель |
||||||||||||
|
|
|
лоидный |
лей |
ного участка к уже имеющимся реп- |
|||||||||||||
|
|
|
коз |
|
|
ликационным вирусным генам и за |
||||||||||||
Мус |
|
ALV, |
Т- и В-клеточ |
мещение |
части |
вирусного |
генома |
|||||||||||
|
|
MuLV, |
ные лимфомы |
фрагментом, несущим иную функцию |
||||||||||||||
|
|
REV, FeLV |
|
|
|
(в частности, онкогеном). В первом |
||||||||||||
Qin |
|
ASV 31 |
Саркома |
|
|
случае |
ретровирус |
сохраняет |
способ |
|||||||||
Raf/Mil/Mht |
|
MuSV |
Карцинома/ |
ность самостоятельно реплицировать |
||||||||||||||
|
|
|
лимфома |
|
|
ся, но приобретает новые свойства |
||||||||||||
Hras |
|
ALV |
Нейробластома |
|||||||||||||||
|
(онкогенность). Типичный представи |
|||||||||||||||||
Kras |
|
F-MuLV |
Эритролейкоз |
тель подобных ретровирусов — вирус |
||||||||||||||
Rel |
|
REV |
В-клеточные |
саркомы |
Рауса. Во |
втором |
случае по |
|||||||||||
|
|
|
лимфомы |
|
|
явление онкогенных |
свойств у ретро- |
|||||||||||
Ros |
|
ASV UR2 |
Саркома |
|
|
|||||||||||||
|
|
|
вируса |
сопровождается |
утратой |
воз |
||||||||||||
Ryk |
|
RPL30 |
|
|
|
|||||||||||||
|
Эритролейкоз/ |
можности самостоятельно реплициро |
||||||||||||||||
Sea |
|
AEV-S13 |
ваться — вирус становится дефект |
|||||||||||||||
|
|
|
саркома |
|
|
ным. Для воспроизведения подобных |
||||||||||||
Sis |
|
SSV |
Глиобластома |
вирусов |
|
необходима |
параллельная |
|||||||||||
Ski |
|
SKV |
Карцинома |
экспрессия вируса-помощника, кото |
||||||||||||||
Src |
|
RSV |
Саркома |
|
|
рый |
|
поставляет |
продукты трансляции |
|||||||||
Yes |
|
Esh и Y73 |
" |
|
|
поврежденных |
генов |
при |
сборке |
ви- |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
104

С х е м а 3.1. Типы структурной организации генома онкогенных
ретровирусов
1 — интактный геном с онкогеном; 2 и 3 — дефектные геномы с онкогеном вместо одного из вирусных генов
русных частиц. Большинство транс |
ных |
незначащими |
последовательно |
|
формирующих ретровирусов дефект |
стями |
(интроны), |
которые |
первона |
ны по репликации и построены по |
чально считываются |
в Р Н К , |
а затем |
|
этому принципу. |
удаляются из нее |
ферментативным |
||
Взависимости от особенностей путем при образовании матричной
структуры вирусного генома онкогены делятся на независимые и слитные. Независимые онкогены транслируют ся в виде отдельного белка, не содер жащего продуктов экспрессии сосед них вирусных последовательностей. При этом независимый онкоген мо жет быть локализован как на конце вирусного генома, так и в середине. Онкогены слитного типа транскриби руются в виде Р Н К , кодирующей онкобелок в составе единой молекулы, несущей аминокислоты структурных вирусных белков. Различные типы структурной организации генома он когенных ретровирусов представлены на схеме 3.1.
Опыты по молекулярной гибриди зации показали, что все без исключе ния вирусные онкогены (v-опс) имеют свои прототипы: протоонкогены (кле точные онкогены — с-опс) в геноме различных клеток. Эти данные яви лись прямым экспериментальным подтверждением ранее высказанной гипотезы о клеточном происхождении вирусных онкогенов.
Протоонкогены, как и большинст во других генов эукариот, имеют ти пичную экзон-интронную структуру, т. е. состоят из кодирующих белковый продукт участков (экзоны), разделен
Р Н К (процесс сплайсинга). В Д Н К всех позвоночных и практически всех других эукариот (дрожжи, насекомые, грибы и пр.) выявлены родственные протоонкогенам по строению и свой ствам гены. Существующая гомология не просто отражает филогенетическое родство организмов, речь идет о не скольких группах древних генов, со хранивших относительно неизмен ную структуру на протяжении дли тельного эволюционного процесса.
Процесс рекомбинации между ретровирусным и клеточным геномами, в результате которого происходит вклю чение определенного клеточного гена в состав генома вируса, называется транедукцией.
Как отмечалось выше, ретровирусы имеют два основополагающих свойства, определяющих своеобразие их репликации: это интеграция в ге ном клетки и обратная транскрипция вирионной Р Н К . Следовательно, ас социация вирусного и клеточного ге номов может происходить либо на уровне Д Н К , т. е. на уровне интегра ции, либо на уровне Р Н К , т. е. на по сттранскрипционном уровне. В на стоящее время механизм включения онкогенов точно не установлен, хотя имеющийся экспериментальный ма-
105
териал позволяет предполагать, что в основном этот процесс реализуется на этапе взаимодействия молекул РНК .
Косвенным свидетельством роли рекомбинации на посттранскрипци онном уровне при формировании onc-содержащих вирусов является су ществование ретровирусов со слож ной структурой или с двумя онкогена ми. Учитывая то обстоятельство, что клеточные последовательности, входя щие в состав этих вирусов, локализо ваны в разных точках клеточного ге нома, следует предположить, что рет ровирусы, состоящие из разных ком понентов, произошли в результате по следовательных интеграции в специ фичные локусы клеточного генома. Естественно, что такая цепь событий маловероятна. Независимо от того, какая из указанных моделей больше соответствует действительности (воз можно, что обе), результатом является включение в состав вирусного генома последовательностей клеточных онко генов без интронов.
Следует подчеркнуть, что все вы шеизложенное может быть отнесено к клеточным генам вообще, т. е. ретровирусы принципиально способны включать в себя различные клеточные последовательности, а не являться пе реносчиками только онкогенов. Пря мым доказательством данного заклю чения являются эксперименты по включению в состав ретровируса в процессе его репликации гена устой чивости к геницитину, введенному в
клетки до вирусной инфекции.
Можно сделать несколько обобще ний, основанных на сравнительном анализе вирусных онкогенов и кле точных протоонкогенов. Главный вы вод заключается в том, что большин ство известных вирусных онкогенов представляют собой каким-либо обра зом измененные варианты нормаль ных клеточных протоонкогенов. Из менения проявляются в различных формах:
• в составе вирусных онкогенов могут быть утрачены определен
ные кодирующие последователь ности, в результате чего вирус ные онкобелки способны утра тить специфичные пептиды, что влечет за собой изменение их ло кализации и функций в клетке;
•изменение свойств онкобелков возможно происходит вследст вие слияния кодирующей облас ти протоонкогенов со структур ными генами ретровируса или определенными гетерологическими последовательностями клеточного генома;
•специфические точечные мута ции, приводящие к замене ами нокислот в активных сайтах онкобелка, могут модифицировать его активность.
Не исключено, что в ряде случаев генетическая информация протоонкогена включается в состав генома онкогенного ретровируса в интактном виде, но в этом случае онкоген будет находиться под контролем сильного вирусного промотора, что может при водить к повышенной и нерегулируе мой его экспрессии.
Взаимодействие ретровируса и кле точного протоонкогена не ограничи вается только процессом трансдукции последнего.
Оказалось, что провирус способен активировать протоонкогены, не включая клеточный генетический ма териал в свою структуру.
Представляется, что факт "внедре ния" вирусной Д Н К в геном клетки хозяина является потенциальным му тагенным фактором. Провирус спосо бен к прямому повреждению генов, локализованных в зоне интеграции. Другая возможность — переход кле точных генов под контроль регуляторных элементов интегрированного в непосредственной близости генома ретровируса — явление инсерционного мутагенеза. Этот феномен в ряде публикаций называется "вставка ви русного промотора". Указанные две возможности относятся к цис-типу взаимодействия провируса и протоон-
J06
когена. Альтернативный вариант инсерционного мутагенеза может реализовываться по транс-типу взаимодей ствия, т. е. независимо от расположе ния провируса и клеточного гена. В этом случае экзогенный провирус продуцирует определенный белковый фактор, способный влиять на актив ность специфических генов — потен циальных онкогенов.
Многочисленные исследования показали, что геномы различных рет ровирусов могут включаться практи чески в любую точку Д Н К клетки, но для некоторых типов провирусов в структуре клеточного генома обнару жены зоны преимущественной инте грации. В ряде случаев подобные уча стки содержат последовательности из вестных протоонкогенов. В зонах ин теграции провируса могут быть распо ложены и не известные ранее потен циальные онкогены. При анализе инсерционного мутагенеза был иденти фицирован ряд новых протоонкоге нов. Информация о клеточных онко генах, обнаруженных при исследова нии ретровирусного инсерционного мутагенеза на различных эксперимен тальных системах, представлена в табл. 3.2.
Основным методическим подходом для идентификации онкогенов, не ас социированных с геномом ретровиру сов, является техника переноса кле точных генов методом трансфекции.
Принцип введения препарата чу жеродной Д Н К в клетку для измене ния ее наследственного потенциала был известен в 40-х годах XX столе тия. Уже тогда было показано, что мо лекулы Д Н К способны переносить ге нетическую информацию и изменять наследственный потенциал клетки. Новый этап в развитии техники трансфекции наступил в 70-е годы, когда был успешно осуществлен пере нос интегрированного провируса в реципиентные клетки, чем была проде монстрирована возможность изучения онкогенов при помощи данного мето да. Попытки трансформировать реципиентные клетки препаратами Д Н К
из различных опухолей оканчивались безуспешно, пока не была усовершен ствована техника переноса ДНК, а в качестве реципиентных клеток не ста ли использовать определенные линии фибробластов грызунов. Обнаружено, что приблизительно 20 % Д Н К тканей всех исследуемых опухолей активны в опытах по трансфекции.
Трансформирующую активность Д Н К из опухоли определяют:
•по способности вызывать фоку сы морфологически измененных клеток.
•на основании роста трансфицированных клеток в полужидком агаре.
•по образованию опухолей у бестимусных мышей, как результат прямой селекции трансформи рованных клеток in vivo.
Вкачестве доказательства переноса трансформирующих последовательно стей из клеток человека могут служить повторяющиеся последовательности семейства alu, характерные для генома человека и содержащиеся в количест ве 300 тыс. копий на клетку. Дальней ший молекулярно-биологический анализ перенесенного фрагмента по зволяет идентифицировать трансфор мирующий ген.
С помощью представленных выше методических подходов (анализ ретро- вирус-ассоциированных генов и трансфекция опухолевой Д Н К ) было выделено подавляющее большинство известных онкогенов.
В последние годы потенциальные онкогенные последовательности вы являют также с помощью ряда прин ципиально новых экспериментальных подходов:
• поиск генов, гомологичных уже известным онкогенам. В этом случае фрагменты различных онкогенов используют в качест ве молекулярных зондов для скрининга репрезентативных библиотек генов исследуемых организмов;
107
Т а б л и ц а |
3.2. Онкогены, |
активируемые |
при инсерции провирусов |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Хромо |
|
|
|
|
Ассоциированные неопла- |
||
Ген/локус |
сомная |
|
|
|
Вирусы |
|||||
локали |
|
|
|
|
зии |
|||||
|
|
|
|
|
|
|
|
|||
|
|
|
зация |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
А. Вирус молоч |
|
|
|
|
|
|
|
|
||
ной железы мы |
|
|
|
|
|
|
|
|
||
шей |
|
|
|
|
|
|
|
|
|
|
Hst-1/kFGF |
|
7 |
|
BR6 |
|
|
Рак молочной железы |
|||
Wnt-1 (Int-1) и |
15 |
|
BALB/cfC3H; BR6; СЗН; |
То |
же |
|
||||
lnt-2 |
|
|
|
|
GR;Grf; |
C3Hf; Mus cervicolor |
|
|
|
|
Int-3 |
|
|
17 |
|
BR6; Czech II |
" " |
|
|
||
Wnt-3 (Int-4) |
11 |
|
BALB/cfC3H; GR |
" " |
|
|
||||
Int-5 |
|
|
9 |
|
BALB/c |
|
|
" |
" |
|
В. Вирусы лей |
|
|
|
|
|
|
|
|
||
коза мышей |
|
|
|
|
|
Пре-В-клеточная |
||||
Ahi-1 |
|
|
10 |
|
Mo-MuLV |
(Abelson) |
||||
Bla-1 |
|
|
9 |
|
F\x-Myc |
|
|
В-клеточная |
пролиферация |
|
Bmi-l/Bup |
|
2 |
|
Ец-Мус |
|
|
То |
же |
|
|
Pal-1 |
|
|
5 |
|
Ец-Мус |
|
|
|
|
|
CSF-1 |
|
3 |
|
BALB/c eco |
Моноцитарный лейкоз |
|||||
Dsi-1 |
|
|
4 |
|
Mo-MuLV |
(rat) |
Т-клеточная |
пролиферация |
||
Evi-2 |
|
|
11 |
|
MuLV (BXH-2) |
Миелолейкоз |
|
|||
Fim-1 |
|
|
13 |
|
F-MuLV |
|
|
|
" |
|
Fim-2/Fms |
|
18 |
|
F-MuLV |
|
|
|
" |
|
|
Fim-3 |
(или |
Evi-1 |
3 |
|
F-MuLV |
|
|
|
" |
|
или |
CB-T) |
|
|
|
|
|
|
|
|
|
Fis-1 |
(или |
Cyl-T) |
7 |
|
F-MuLV |
|
|
|
|
|
Fli-1 |
|
|
9 |
|
F-MuLV |
|
|
Эритролейкоз |
||
Gin-1 |
|
|
19 |
|
Gross A |
|
|
Т-клеточная |
пролиферация |
|
Lck |
|
|
4 |
|
Mo-MuLV |
|
То же |
|
||
Mlvi-1 (или Pvt, |
15 |
|
AKR, AKXD, Mo-MuLV (rat) |
" |
" |
|
||||
Mis-1 |
или |
|
|
|
|
|
|
|
|
|
RMO-int-I) |
|
|
|
|
|
|
|
|
|
|
Mlvi-2, -3, -4 |
15 |
|
Mo-MuLV |
(rat) |
it |
11 |
|
|||
|
|
|
|
|||||||
Myb |
|
|
10 |
|
Mo-MuLV |
(Abelson) |
Миелолейкоз |
|
||
|
|
|
|
|
Cas-Br-M |
|
NFS-60 клеточная линия |
|||
Мус |
|
|
15 |
|
AKR; AKXD, Gross A; |
Т-клеточная |
пролиферация |
|||
|
|
|
|
|
MCF247; MCF69L1; Mo- |
|
|
|
||
|
|
|
|
|
MuLV (rat); Soule; Ец-Pim-1 |
|
|
|
||
Nmyc-1 |
|
12 |
|
MCF247; Mo-MuLVEu-Pim-! |
То же |
|
||||
Pim-1 |
|
|
17 |
|
AKR; AKXD; MCF247; |
|
|
|
||
|
|
|
|
|
MCF1233; |
MCF69L1 |
Не-Т-клеточная пролифе |
|||
|
|
|
|
|
AKXD |
|
|
рация |
|
|
|
|
|
|
|
ДМо-MuLV + SV |
В-лимфобластный лейкоз |
||||
Pim-2 |
|
|
17 |
|
AKDX; |
Mo-MuLV |
Т-клеточная |
пролиферация |
||
Hras |
|
|
7 |
|
Mo-MuLV |
|
То же |
|
||
Kras |
|
|
6 |
|
F-MuLV |
|
|
Миелолейкоз |
|
|
Sic-1 |
|
|
9 |
|
Cas-Br-E |
MuLV |
Не В-клеточная, не Т-кле |
|||
|
|
|
|
|
|
|
|
точная пролиферация |
||
Spi-1 |
|
|
2 |
|
SFFV |
|
|
Эритролейкоз |
||
P53 |
|
|
11 |
|
F-MuLV; |
SFFV |
|
|
|
|
|
|
|
|
|
Abelson |
MuLV |
Лимфолейкоз |
|||
Tpl-I/Els-1 |
|
9 |
|
Mo-MuLV |
|
Т-клеточная |
пролиферация |
|||
Vin-1 |
|
|
6 |
|
RadLV |
|
|
То |
же |
|
С. Ретровирусы |
|
|
|
|
|
|
|
|
||
птиц |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
108
Продолжение
|
|
|
|
|
Хромо |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ген/локус |
|
|
сомная |
|
|
|
|
Вирусы |
|
|
|
Ассоциированные |
неопла- |
||||||||||
|
|
локали |
|
|
|
|
|
|
|
|
|
зии |
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
зация |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Bic |
|
|
|
|
|
|
|
UR2AV + RAV-2 |
|
|
|
Лимфомы |
|
|
|
|
|
||||||
Erbb |
|
|
|
|
|
|
|
RAV-1 |
|
|
|
|
|
Эритробластозы |
|
|
|
||||||
Myb |
|
|
|
|
|
|
|
RAV-1; EU-8;UR2AV |
+ |
|
|
Лимфомы |
|
|
|
|
|
||||||
Мус |
|
|
|
|
|
|
|
RAV-2 |
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
ALV; |
REV (CSV); RPV |
|
|
В-клеточная |
лимфома |
|
|
|||||||||
|
|
|
|
|
|
|
|
RPV |
|
|
|
|
|
|
Аденокарцинома |
|
|
|
|||||
Nov |
|
|
|
|
|
|
|
REV |
|
|
|
|
|
|
Т-клеточная |
лимфома |
|
|
|||||
|
|
|
|
|
|
|
MAV1 |
|
|
|
|
|
Миелобластоз |
|
|
|
|
||||||
Hras |
|
|
|
|
|
|
|
MAV |
|
|
|
|
|
Нефробластома |
|
|
|
|
|||||
Blym |
|
|
|
|
|
|
|
ALV |
|
|
|
|
|
|
В-клеточная |
лимфома |
|
|
|||||
Rel |
|
|
|
|
|
|
|
ALV |
|
|
|
|
|
|
То же |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
• |
выявление |
генов, |
|
активирован |
опухолевую? |
Несколько |
групп |
кос |
|||||||||||||||
|
ных (или подавленных) с помо |
венных |
фактов |
свидетельствуют |
в |
||||||||||||||||||
|
щью |
различных |
модификаций |
пользу того, что это величина огра |
|||||||||||||||||||
|
принципа |
"вычитания |
|
генов". |
ниченная. |
|
|
|
|
|
|
|
|
||||||||||
|
Для |
этого |
осуществляют |
отбор |
Во-первых, несмотря на активное |
||||||||||||||||||
|
комплементарных Д Н К (кДНК), |
исследование |
с |
|
использованием |
|
со |
||||||||||||||||
|
специфически |
гибридизующих- |
временных изощренных молекулярно- |
||||||||||||||||||||
|
ся с Р Н К из определенных типов |
биологических приемов, |
в |
последние |
|||||||||||||||||||
|
опухолей, |
|
|
но |
не |
взаимодейст |
годы число вновь открываемых онко |
||||||||||||||||
|
вующих |
с |
Р Н К |
|
аналогичной |
генов резко сократилось. |
|
|
|
|
|
||||||||||||
|
нормальной ткани или доброка |
Во-вторых, один и тот же онкоген |
|||||||||||||||||||||
|
чественной |
опухоли. Разработа |
определяет трансформирующие свой |
||||||||||||||||||||
|
ны |
различные |
модификации |
ства |
разных ретровирусов. |
|
|
|
|
||||||||||||||
|
данного |
принципа: |
разностная |
В-третьих, во многих новых онко- |
|||||||||||||||||||
|
гибридизация, |
дифференциаль |
генных вирусах встречаются уже опи |
||||||||||||||||||||
|
ный |
дисплей, |
сравнительный |
санные онкогены. |
|
|
|
|
|
||||||||||||||
|
параллельный |
скрининг |
иммо |
В-четвертых, при анализе транс |
|||||||||||||||||||
|
билизованных |
|
|
|
фрагментов |
формирующих |
потенций |
Д Н К |
раз |
||||||||||||||
|
кДНК из нормальной и опухоле |
личных опухолей методом трансфек- |
|||||||||||||||||||||
|
вой ткани и др. Сочетанием этих |
ции в большинстве случаев идентифи |
|||||||||||||||||||||
|
методов является скрининг экс- |
цируется ограниченный круг одних и |
|||||||||||||||||||||
|
прессирующих |
белки |
библиотек |
тех же онкогенов. |
|
|
|
|
|
||||||||||||||
|
к Д Н К сыворотками |
от онколо |
В |
настоящее |
|
время |
известно |
|
не |
||||||||||||||
|
гических |
больных. |
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
многим более 100 различных протоон- |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
когенов, изменения структуры или ги |
|||||||||||
Следует отметить, что, несмотря на |
перэкспрессия |
которых |
приводят |
к |
|||||||||||||||||||
колоссальные |
|
возможности |
приме |
продукции онкобелков. Почти все из |
|||||||||||||||||||
няемых методов, число новых онкоге |
вестные |
онкогены кодируют |
белки, |
||||||||||||||||||||
нов, идентифицированных с их помо |
участвующие |
в |
важнейших |
процессах |
|||||||||||||||||||
щью, относительно невелико. |
|
|
жизнедеятельности клетки. Считается, |
||||||||||||||||||||
Сколько |
всего онкогенов? |
Или, |
что число идентифицированных онко |
||||||||||||||||||||
другими словами, каково число нор |
генов (около 100 из примерно 50 тыс. |
||||||||||||||||||||||
мальных клеточных |
генов, |
которые |
функциональных |
генов |
клетки) |
при |
|||||||||||||||||
при |
определенных |
условиях |
могут |
ближается |
к |
естественному |
пределу, |
||||||||||||||||
переводить |
нормальную |
клетку в |
определяемому |
|
ключевыми |
точками |
|||||||||||||||||
109
Т а б л и ц а 3.3. Локализация протоонкогенов в хромосомах человека
Хромосома |
Ген |
Хромосома |
Ген |
|
|
|
|
1 |
ECK |
10q24 |
HOX11 |
1 |
EEK |
llpl2-pll.2 |
SPI1 |
1 |
ERK |
llpl3 |
RBTNL1 |
1 |
TPR |
llpl3 |
WT1 |
1р13 |
NRAS |
1 lpl5 |
RBTN1 |
1р32 |
BLYM |
llpl5.5 |
HRAS |
1р32 |
MYCLl |
llql3 |
BCL1 |
1р32 |
TALI |
llql3 |
INT2 |
1р32-р31 |
JUN |
liql'3 |
MEN1 |
1рЗЗ |
SIL |
llql3 |
SEA |
1рЗЗ-34 |
TIE |
llql3.3 |
HSTF1 |
1р34 |
MPL |
I lq23 |
MLL |
1р35-р32 |
LCK |
llq23 |
PLZF |
1р36.2-р36.1 |
PGR |
Ilq23-q24 |
ERG В |
1q22-q24 |
SKI |
llq23.3 |
ETSJ |
lq23 |
PBX1 |
llq23.3-qter |
CBL |
1q23-q24 |
TRK |
Mq24 |
FLU |
1q23-q31 |
TRKB |
12pl2.1 |
KRAS2 |
1q23-q31 |
TRKC |
12pl3 |
HST2 |
1q24-q25 |
ABL2 (ARG) |
12ql3 |
HER3 |
2pl3-pl2 |
REL |
12ql3 |
WNT1 |
2p24.1 |
MYCN |
12ql3-ql4.3 |
GLI1 |
2p25 |
ODC1 |
12q22 |
BTG1 |
2ql4-q21 |
HIS1 |
13ql2 |
FLTl |
2ql4-q21 |
LCO |
13ql2 |
FLT3/FLK2 |
2q33-qter |
INHA |
13ql4.2 |
RBI |
3p24.1-p22 |
THRB |
14q24.3 |
FOS |
3p25 |
RAF1 |
14q32 |
АКТ |
3p25-p26 |
VHL |
14q32.3 |
ELK2 |
3q22 |
RYK |
15 |
CSK |
3q26 |
EVI |
15ql3-q21 |
LTK |
4q21 |
AF4 |
15q21 |
PML |
4 |
FLK1/KDR |
15q25-q26 |
FPS/FES |
4qll-q21 |
KIT |
16q |
С MAR/CAR |
5/17/18 (elements) |
TRE |
16q22.1 |
UVO |
5q21 |
FER |
17pl3.1 |
TPS3 |
5q21 |
MCC |
17ql 1.2 |
EV12B |
5q21-q22 |
A PC |
17ql 1.2 |
NF1 |
5q31.3-q33.2 |
FGFA |
17qll.2-ql2 |
THRA1 |
5q33.3-q34 |
CSF1R |
17ql2-q21 |
BRCA1 |
5q35 |
FLT4 |
17q21 |
PHB |
6p21 |
PIM1 |
17q21 |
RARA |
6p23-p22.3 |
FIM1 |
17q21-q22 |
HER2 |
6q21 |
FYN |
17q21-q22 |
WNT3 |
6q21-q22 |
ROS1 |
17q21.3 |
NME1, NME2 |
6q22-q23 |
MYB |
18q21 |
BCL2 |
6p23 |
ВЕК |
18q21 |
DCC |
6q24-q27 |
MAS |
18q21.3 |
YES1 |
7pl3-pl2 |
EGFR |
19pl3.2 |
LYL1 |
7pl5 |
MYCLK1 |
19pl3.2 |
JUNB |
7q31 |
MET |
19pl3.2 |
JUND |
7q31 |
WNT2 |
19pl3.2 |
VAV |
7q32-q36 |
EPHT |
19pI3.3 |
ENL |
7q33-q36 |
RAFB1 |
19cen-pl3.2 |
MEL |
8pl2 |
FLT2 |
19ql3.1 |
BCL3 |
8qll-ql2 |
MOS |
19ql3.1 |
UFO |
8ql3-qter |
LYN |
20qll |
ИСК |
8q22 |
MYB LI |
20ql3.3 |
SRC |
8q22 |
ETO |
21q22 |
AM LI |
8q24 |
MYC |
21q22.3 |
ERG |
8q24 |
PVTI |
21q22.3 |
ETS2 |
8q24.1 |
NOV |
22ql 1.2 |
BCR |
9q22 |
AF9 |
22ql2 |
EWS |
9q34.1 |
ABL1 |
22ql2 |
NF2 |
9q34 |
CAN |
22ql2.3-ql3.1 |
PDGFB |
9q34 |
SET |
Xpll.2 |
ELK1 |
9q34 |
TAL2 |
Xpll.2 |
RAFA1 |
9q34 |
TAN1 |
Xpll.3-pll.23 |
TIMP |
10pll.2 |
EST |
Xql3 |
MYBL2 |
10qll.2 |
RET |
Xq27 |
DBL |
|
|
|
|
Т а б л и ц а 3.4. Функции онкобелков
Класс 1 Факторы роста PDGF/SIS, INT2, HST1, WNT1/WNT3
Класс 2 Тирозинкиназы
|
|
Тирозинкиназы рецепторные |
||
|
|
ЕРН, ELK, EGFR/ERBB, FMS, |
||
|
|
KIT, MET, |
H E R / N E U , RET, |
|
|
|
ROS, SEA, TRK |
||
|
|
Тирозинкиназы нерецепторные |
||
|
|
SRC (SRC-родственные киназы |
||
|
|
FGR, FYN, HCK, LCK, LYN/ |
||
|
|
SYN, YES), |
ABL, CSK/CYL, |
|
|
|
FPS/FES |
|
|
Класс |
3 |
Некиназные |
рецепторы |
|
|
|
MAS, MPL |
|
|
Класс |
4 |
G-белки, ассоциированные с мем |
||
|
|
браной |
|
|
|
|
HARAS, |
KRAS, NRAS, GSP |
|
Класс |
5 |
Белки, |
связывающие комплексы |
|
|
|
Rho/Rac |
|
|
|
|
BCR, DBL |
|
|
Класс |
6 |
Цитоплазматические сериновые |
||
|
|
киназы |
|
|
|
|
BCR, MOS, PIM1, RAF/MIL |
||
Класс |
7 |
Цитоплазматические регуляторы |
||
|
|
BCL1, CRK |
|
|
Класс |
8 |
Факторы транскрипции |
||
|
|
BCL3, ETS, EVI1, FOS (FOS- |
||
|
|
родственные белки FOSB, FRA1, |
||
|
|
FRA2), J U N |
(JUN-родственные |
|
|
|
белки J U N B , J U N D ) , MYB, |
||
|
|
MYC (MYC-родственные белки |
||
|
|
MYCL, MYCN), REL, TALI, SKI |
||
Класс |
9 |
Негативный регулятор апоптоза |
||
|
|
BCL2 |
|
|
известных биохимических процессов в клетках.
Естественно, что наиболее активно исследовали протоонкогены человека, которые в настоящее время обнаруже ны практически на всех хромосомах (табл. 3.3).
Заключая данный раздел, следует подчеркнуть два основополагающих биологических факта, связанных с он когенами:
открытие определенных генов, ассоциированных с индукцией и развитием процесса опухолеобразования; открытие феномена "консерва
тивности" протоонкогенов, т. е.
их присутствия в геноме эволюционно отдаленных организ мов.
Из накопленного эксперименталь ного материала следует, что протоон когены появились до разделения эво люционных путей насекомых и хордо вых (более 800 млн лет назад). По-ви димому, формирование этих генов связано с возникновением многокле точных организмов. Об этом свиде тельствуют данные об обнаружении сходных последовательностей у такого примитивного организма, как дрож жи, а также отсутствие информации о существовании аналогичных элемен тов в геноме одноклеточных.
Научная классификация онкогенов в настоящее время отсутствует. Гены распределены в соответствии с извест ными функциями белковых продук тов, кодируемых их клеточными гомо логами — протоонкогенами. Примеры подобного распределения ряда наибо лее известных онкобелков представле ны в табл. 3.4.
Присутствие онкогенов в клетках различных видов имеет принципиаль ное значение для исследователей их свойств, так как позволяет проводить эксперименты на различных модель ных системах. В результате многочис ленных экспериментов были установ лены основные молекулярные меха низмы, в результате реализации кото рых нормальный клеточный ген — протоонкоген — приобретает свойства онкогенов.
3.1.2. Механизмы активации протоонкогенов
Процесс превращения нормально го протоонкогена в онкоген называет ся активацией. Известно 3 основных механизма активации протоонкогенов (схема 3.2):
•мутации в первичной структуре протоонкогена;
•амплификация протоонкогена (увеличение числа копий гена в
ДН К клетки);
111
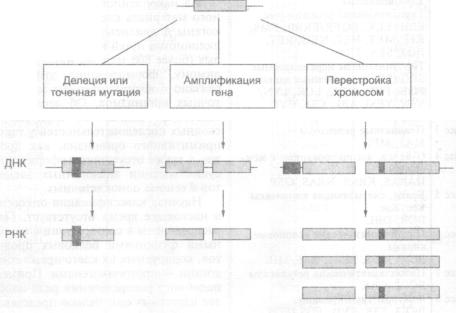
С х е м а 3.2. Основные молекулярные механизмы активации протоонкогенов
Кодирующая
последовательность
|
Повышенная |
Присоединение к |
|
Продукция |
энхансеру или |
||
продукция |
|||
измененного |
активному гену: |
||
нормального |
|||
белка |
повышенная продукция |
||
белка |
|||
|
белка |
||
|
|
•перестройка генома (хромосом) ции затрагивают 12 или 61 кодон, что
клетки, |
затрагивающая |
структу |
выражается |
в |
замене |
в |
белке ras |
|||||||
ру протоонкогена. |
|
|
|
|
(p21ras) |
определенных |
аминокислот. |
|||||||
|
|
|
|
|
|
|
|
Подлинный белок p21ras обладает гуа- |
||||||
Ниже |
представлены |
наиболее |
ха |
нозинтрифосфатазной |
акти вностью: |
|||||||||
рактерные |
примеры |
активации прото |
гидролизует |
|
гуанозинтрифосфат |
|||||||||
онкогенов по каждому из известных |
(ГТФ) до гуанозиндифосфата (ГДФ) и |
|||||||||||||
механизмов, обнаруженных |
при |
ана |
свободного фосфата. Мутации Glyl2 |
|||||||||||
лизе некоторых неоплазий человека. |
или Gln61 |
приводят к резкому сниже |
||||||||||||
Мутации. В результате мутаций в |
нию этой активности. Появление в |
|||||||||||||
структуре |
гена |
изменяется |
кодирую |
белке p21ras любой аминокислоты (за |
||||||||||
щий белок, что отражается на его |
исключением пролина) |
вместо глици |
||||||||||||
свойствах. |
Наиболее |
убедительные |
и |
на в 12-м положении придает ему |
||||||||||
обширные |
экспериментальные |
дан |
трансформирующие свойства. В |
ком |
||||||||||
ные о роли специфических мутаций в |
плексе с ГДФ p21ras является неак |
|||||||||||||
активации клеточных онкогенов |
были |
тивным, а при ассоциации с ГТФ он- |
||||||||||||
получены |
при |
сравнительном анализе |
кобелок |
приобретает активную |
кон |
|||||||||
генов семейства ras. У разных пред |
фигурацию |
и |
стимулирует связываю |
|||||||||||
ставителей |
этой группы |
генов |
мута |
щиеся с |
ним |
белки. |
В |
нормальных |
||||||
112
клетках баланс между активной и пас сивной формами белка ras строго ре гулируется (см. ниже). Вследствие онкогенных мутаций белок остается в комплексе с ГТФ, сохраняется в пер манентно активной форме, нарушает нормальное прохождение сигналов, что в конечном итоге приводит к трансформации клетки. Активирую щие мутации в протоонкогенах семей ства ras обнаруживаются в широком спектре опухолей человека. При этом доля опухолей с такими мутациями варьирует от единичных случаев при раке молочной железы и раке желудка до 95 % случаев при раке поджелудоч ной железы.
Амплификация. Основным резуль татом другого механизма активации онкогенов — амплификации — явля ется аномально высокая продукция кодируемого белка в клетках (см. схе му 3.2).
фикация онкогена коррелирует с оп ределенным новообразованием и мо жет служить в качестве молекулярного маркера при диагностике и определе нии прогноза заболевания.
Транслокация. Превращение про тоонкогена в активированный онко ген может являться следствием раз личных структурных перестроек кле точного генома. Обмен генетического материала осуществляется как между гомологичными, так и между негомо логичными хромосомами. Как прави ло, этот процесс представляет собой сбалансированный реципрокный ме ханизм (т. е. взаимный, равноценный обмен фрагментами генома), но воз можна и потеря Д Н К в одной или обоих точках рекомбинации. В ре зультате подобных событий гены (протоонкогены, в частности), лока лизованные в зоне реаранжировки ге нома, могут претерпевать драматиче
Вразличных опухолях человека ские изменения своей структуры: ут
идентифицирована амплификация ря |
рату части генетической информации, |
||||||||||||
да известных онкогенов. В большин |
образование |
химерных |
генов, |
коди |
|||||||||
стве случаев в опухолях человека в ам- |
рующих |
гибридные |
белки, попадание |
||||||||||
плифицированном |
состоянии |
встре |
в зону регуляторных элементов других |
||||||||||
чаются |
онкогены |
семейства |
MYC. |
генов |
с |
последующим |
нарушением |
||||||
Максимальное число копий демонст |
нормальной |
регуляции |
экспрессии. |
||||||||||
рирует онкоген NMYC в случае ней- |
Практически |
все |
упомянутые |
меха |
|||||||||
робластом и ретинобластом — до 200 |
низмы |
качественного изменения он |
|||||||||||
копий на гаплоидный геном клетки. В |
когенов в результате перестройки кле |
||||||||||||
подобных |
случаях |
умножение |
гена |
точной Д Н К |
описаны для различных |
||||||||
может быть обнаружено цитогенети- |
опухолей |
человека. |
|
|
|
||||||||
ческими методами в виде удлиненных |
Наиболее ярким примером специ |
||||||||||||
областей |
хромосом |
с нетипичным |
фической активации онкогенов в ре |
||||||||||
распределением полос при окраске. |
зультате |
хромосомной |
транслокации |
||||||||||
Обычно же амплификация онкогенов |
является |
образование |
филадельфий |
||||||||||
не |
превышает нескольких |
десятков |
ской хромосомы (Ph1) при CML. |
||||||||||
копий и выявляется более чувстви |
Классическая |
реципрокная трансло |
|||||||||||
тельными |
|
молекулярно-биологиче- |
кация 9;22 регистрируется в 95 % слу |
||||||||||
скими методами. В настоящее время |
чаев данного лейкоза. На молекуляр |
||||||||||||
нет строгих экспериментальных дока |
ном уровне это выражается в переносе |
||||||||||||
зательств того, что именно увеличение |
протоонкогена ABL на хромосому 22 в |
||||||||||||
числа копий |
определенных |
протоон |
зону местонахождения гена BCR. Об |
||||||||||
когенов индуцирует появление |
опухо |
разуется химерный ген BCR-ABL, ко |
|||||||||||
ли. Наоборот, есть серьезные основа |
торый в результате последующей пе |
||||||||||||
ния предполагать, что этот процесс |
рестройки локализуется |
в аномальной |
|||||||||||
имеет вторичную природу и отражает |
хромосоме Ph1. Белковый продукт хи |
||||||||||||
селективные |
преимущества |
|
одних |
мерного |
гена представляет |
собой |
|||||||
клеток перед другими. Но несомнен |
слитную |
молекулу |
p210bcr-abl. |
|
|||||||||
но, |
что в некоторых случаях ампли- |
Другим ярким примером специфи- |
|||||||||||
|
|
|
|
|
|
|
|||||||
8 - 7908 Д. Г. Заридзе |
|
|
|
|
|
|
|
|
|
|
113 |
||
ческой |
генной транслокации является |
хромосом, |
вызывающие развитие опу |
||||||||||||||||||||
лимфома Беркитта (ЛБ, BL). В случае |
холей, |
необязательно должны затраги |
|||||||||||||||||||||
эндемической |
формы |
BL |
опухолевые |
вать лишь протоонкогены. Изменени |
|||||||||||||||||||
клетки |
содержат |
Д Н К |
вируса |
Эп- |
ям могут подвергаться и другие после |
||||||||||||||||||
штейна—Барр (EBV). При спорадиче |
довательности |
Д Н К , |
например гены, |
||||||||||||||||||||
ских формах этого заболевания вирус |
кодирующие |
белки-мишени |
|
продук |
|||||||||||||||||||
ный геном выявляется в 25 % случаев, |
тов онкогенов. |
|
|
|
|
|
|
|
|||||||||||||||
хотя и в этом случае 75 |
% больных се- |
|
|
|
|
|
|
|
|||||||||||||||
Из |
имеющихся |
на |
сегодняшний |
||||||||||||||||||||
ропозитивны |
по |
EBV. |
|
Наблюдаемые |
|||||||||||||||||||
|
день |
экспериментальных |
|
данных |
|||||||||||||||||||
хромосомные |
перестройки |
в |
основ |
|
|||||||||||||||||||
сравнительного |
анализа |
онкогенов |
в |
||||||||||||||||||||
ном |
являются |
реципрокными |
транс |
||||||||||||||||||||
различных |
опухолях |
следует |
принци |
||||||||||||||||||||
локациями |
между |
хромосомами |
8 и |
||||||||||||||||||||
пиальный вывод: |
|
|
|
|
|
|
|||||||||||||||||
14, |
но |
примерно |
в |
10 % случаев |
на |
|
|
|
|
|
|
||||||||||||
• |
во всех детально изученных слу |
||||||||||||||||||||||
блюдаются |
альтернативные трансло |
||||||||||||||||||||||
|
чаях |
онкогены, |
ассоциирован |
||||||||||||||||||||
кации: 2;8 или 8;22. На генном уровне |
|
||||||||||||||||||||||
|
ные с определенным типом опу |
||||||||||||||||||||||
указанные |
изменения |
в |
Д Н К |
клеток |
|
||||||||||||||||||
|
холи, |
отличались |
по |
своей |
|||||||||||||||||||
выражаются в перемещении протоон |
|
||||||||||||||||||||||
|
структуре |
от |
соответствующих |
||||||||||||||||||||
когена MYC, локализованного на хро |
|
||||||||||||||||||||||
|
протоонкогенов |
|
нормальной |
||||||||||||||||||||
мосоме 8, в область генов, кодирую |
|
|
|||||||||||||||||||||
щих |
тяжелые |
цепи |
иммуноглобули |
|
ткани. |
|
|
|
|
|
|
|
|
||||||||||
нов, лямбда-легкие цепи и гама-легкие цепи на |
|
|
|
|
|
|
|
|
|
|
|||||||||||||
14, 2 и 22-й хромосомах соответствен |
3.1.3. Онкогены в системе |
|
|||||||||||||||||||||
но. В большинстве исследованных |
передачи |
|
сигналов |
|
|
|
|||||||||||||||||
случаев основной транслокации |
(8; 14) |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
происходят изменения гена MYC, не |
Распределение онкогенов |
по огра |
|||||||||||||||||||||
затрагивающие его |
кодирующую |
об |
ниченному числу групп генов с близ |
||||||||||||||||||||
ласть: |
утрачиваются |
или |
нарушаются |
кими свойствами позволяет предполо |
|||||||||||||||||||
регуляторные |
структуры |
гена. |
|
Экс |
жить, что спектр молекулярных меха |
||||||||||||||||||
прессия функционального |
белка myc |
низмов канцерогенеза достаточно ог |
|||||||||||||||||||||
является |
обязательным |
|
признаком |
раничен, |
а процессы |
малигнизации, |
|||||||||||||||||
клеток |
BL. |
При |
"альтернативных" |
ассоциированные |
с |
|
онкогенами, |
||||||||||||||||
транслокациях (2;8, 8;22) протоонко- |
должны иметь общие закономерности. |
||||||||||||||||||||||
ген MYC сохраняет свое местонахож |
Естественно, |
что |
для |
понимания |
|||||||||||||||||||
дение на 8-й хромосоме, но происхо |
механизмов |
реализации |
потенциала |
||||||||||||||||||||
дит перенос участка генов иммуногло |
онкогенов |
необходима |
информация о |
||||||||||||||||||||
булинов с 2-й и 22-й хромосом в об |
роли исходных протоонкогенов в нор |
||||||||||||||||||||||
ласть гена MYC. Взаимное расположе |
м а л ь н о й клетке. |
|
|
|
|
|
|
|
|||||||||||||||
ние перемещенных генов может иметь |
С точки зрения современной био |
||||||||||||||||||||||
как одинаковую (голова — хвост), так |
логии |
гармоничное |
функционирова |
||||||||||||||||||||
и обратную (голова |
— |
голова) |
ориен |
ние многоклеточного организма опре |
|||||||||||||||||||
тацию. Трансформирующий потенци |
|||||||||||||||||||||||
деляется |
способностью |
его |
клеток |
||||||||||||||||||||
ал |
онкогена |
MYC |
реализуется |
как |
|||||||||||||||||||
взаимодействовать |
между |
собой, |
а |
||||||||||||||||||||
следствие |
различных |
молекулярных |
|||||||||||||||||||||
также |
их адекватной |
реакцией |
на из |
||||||||||||||||||||
механизмов, |
но |
решающим |
общим |
||||||||||||||||||||
менение окружающей среды. "Органа |
|||||||||||||||||||||||
признаком всех лимфом подобного ти |
|||||||||||||||||||||||
ми чувств" клетки являются поверхно |
|||||||||||||||||||||||
па является ассоциация локусов прото |
|||||||||||||||||||||||
стные рецепторы, которые восприни |
|||||||||||||||||||||||
онкогена MYC и генов цепей иммуног |
|||||||||||||||||||||||
мают |
внеклеточные |
факторы |
и соот |
||||||||||||||||||||
лобулинов в составе одной хромосомы. |
|||||||||||||||||||||||
ветствующим |
образом |
"информируют" |
|||||||||||||||||||||
Предполагается, |
что |
|
транслокация |
||||||||||||||||||||
|
клетку. Специфическое |
взаимодейст |
|||||||||||||||||||||
происходит |
в |
период дифференциров |
|||||||||||||||||||||
вие внешних факторов и |
мембранных |
||||||||||||||||||||||
ки, |
сопровождаемой |
реаранжировкой |
|||||||||||||||||||||
рецепторов инициирует процесс пере |
|||||||||||||||||||||||
генов |
иммуноглобулинов. |
|
|
|
|||||||||||||||||||
|
|
|
дачи сигналов: |
"импульс", возникаю |
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
Следует |
отметить, |
что |
перестройки |
щий на клеточной мембране, усилива- |
|||||||||||||||||||
П4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ется и передается внутрь клетки по |
розинсвязывающий домен (РТВ), ко- |
||||||||||||||||||||||
определенным |
цепям |
передачи сигна |
торые узнают участок белка, несущий |
||||||||||||||||||||
ла. Некоторые из сигналов направля |
фосфотирозин; SHЗ-домен взаимо |
||||||||||||||||||||||
ются в клеточное ядро и запускают |
действует |
с |
обогащенными |
пролином |
|||||||||||||||||||
экспрессию |
определенных |
генов. От |
элементами белка; РН-домен узнает |
||||||||||||||||||||
вет клетки на сигнальный стимул мо |
мембранные фосфолипиды и др. |
|
|||||||||||||||||||||
жет иметь различное проявление: де |
Белковые продукты многих прото- |
||||||||||||||||||||||
ление клетки (или, наоборот, останов |
онкогенов |
представляют |
собой важ |
||||||||||||||||||||
ка этого процесса), дифференциация, |
нейшие |
компоненты |
проведения |
и |
|||||||||||||||||||
секреция гормонов и т. д. |
Системы |
контроля сигналов в клетке. В даль |
|||||||||||||||||||||
передачи сигналов в клетке имеют |
нейшем |
эти |
процессы |
рассматрива |
|||||||||||||||||||
очень сложную организацию и, не |
ются на примерах наиболее изучен |
||||||||||||||||||||||
смотря |
на |
активные |
исследования, |
ных |
прототипных |
белков, |
кодируе |
||||||||||||||||
имеющаяся |
информация |
достаточно |
мых основными классами протоон- |
||||||||||||||||||||
фрагментарна. |
|
|
|
|
|
|
|
когенов. Общая схема передачи сиг |
|||||||||||||||
В упрощенном виде механизм пе |
налов |
от |
различных |
внеклеточных |
|||||||||||||||||||
редачи сигналов в клетке представляет |
лигандов |
внутрь |
клетки |
представлена |
|||||||||||||||||||
собой |
прямое |
взаимодействие специ |
на схеме 3.3. |
|
|
|
|
|
|
|
|||||||||||||
фических белков в строго определен |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
ной последовательности. |
Ряд ключе |
3.1.3.1. |
Сигналы, |
|
|
|
|
||||||||||||||||
вых белков может быть задействован в |
|
|
|
|
|||||||||||||||||||
стимулированные PDGF |
|
||||||||||||||||||||||
нескольких сигнальных путях. Основ |
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
ными |
участниками |
цепей |
передачи |
Наиболее изученным |
представите |
||||||||||||||||||
сигналов в клетках являются различ |
лем |
протоонкогенов, |
относящихся |
к |
|||||||||||||||||||
ные типы киназ, а также адапторные |
классу факторов роста, является фак |
||||||||||||||||||||||
молекулы, которые не обладают фер |
тор роста тромбоцитов — прототип |
||||||||||||||||||||||
ментативной |
активностью. |
Адаптеры |
вирусного онкогена sis (PDGF/sis). |
||||||||||||||||||||
активируются |
специфическими кина- |
Этот фактор стимулирует рост и под |
|||||||||||||||||||||
зами и приобретают способность об |
вижность клеток соединительной тка |
||||||||||||||||||||||
разовывать комплексы с другими со |
ни (фибробластов и клеток гладких |
||||||||||||||||||||||
ставляющими |
процесса передачи |
сиг |
мышц), хотя |
способен |
воздействовать |
||||||||||||||||||
нала. Фактически основными молеку |
и на клетки другой природы (эндоте- |
||||||||||||||||||||||
лярными принципами, на которых ба |
лиальные |
клетки |
и |
нейроны). P D G F |
|||||||||||||||||||
зируется |
механизм |
передачи |
сигналов |
продуцируется |
|
главным |
образом |
||||||||||||||||
в клетке, являются специфическая ас |
тромбоцитами |
и |
является |
основным |
|||||||||||||||||||
социация белков и их фосфорилиро- |
ростовым фактором сыворотки. Мо |
||||||||||||||||||||||
вание |
|
(или |
дефосфорилирование). |
лекула |
P D G F представляет |
собой ди- |
|||||||||||||||||
Фосфорилирование |
белков-мишеней |
мерный |
комплекс |
ассоциированных |
|||||||||||||||||||
ведет к мгновенному изменению их |
дисульфидными |
связями |
полипептид |
||||||||||||||||||||
конфигурации и свойств. Баланс меж |
ных |
цепей |
А |
и |
В. |
Как |
гомодимеры |
||||||||||||||||
ду фосфорилированием и дефосфори- |
( P D G F АА, |
P D G F |
ВВ), |
так и гетеро- |
|||||||||||||||||||
лированием |
|
определяет |
|
передачу |
димер ( P D G F АВ) действуют на клет |
||||||||||||||||||
внутриклеточных сигналов в норме. |
ки-мишени через соответствующие |
||||||||||||||||||||||
Физическое |
взаимодействие |
сиг |
поверхностные рецепторы |
тирозино- |
|||||||||||||||||||
вых протеинкиназ (а- и β-рецепторы). |
|||||||||||||||||||||||
нальных |
молекул |
осуществляется за |
|||||||||||||||||||||
Известно, что подавляющее большин |
|||||||||||||||||||||||
счет определенных универсальных по |
|||||||||||||||||||||||
ство |
киназных рецепторов |
активиру |
|||||||||||||||||||||
следовательностей, |
входящих |
в струк |
|||||||||||||||||||||
ются |
под |
действием |
лигандов в виде |
||||||||||||||||||||
туру |
этих |
белков. |
Практически |
все |
|||||||||||||||||||
димерных |
|
комплексов. |
На |
примере |
|||||||||||||||||||
белки-компоненты |
цепей |
передачи |
|
||||||||||||||||||||
P D G F показано, что лиганд одновре |
|||||||||||||||||||||||
сигналов |
имеют |
определенное сочета |
|||||||||||||||||||||
менно |
взаимодействует с двумя моле |
||||||||||||||||||||||
ние соответствующих доменов. К |
по |
||||||||||||||||||||||
кулами рецептора, что приводит к ак |
|||||||||||||||||||||||
добным |
структурам |
относятся: |
SH2 |
||||||||||||||||||||
тивации последнего. Димеризация ре- |
|||||||||||||||||||||||
(src-homology 2) |
— домен и |
фосфоти- |
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
8 * |
115 |
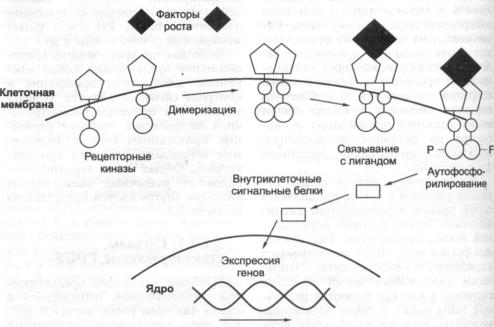
С х е м а 3.3. Общая схема передачи сигналов от внеклеточных лигандов внутрь
клетки
цепторов — ключевое событие в ин дукции и последующей передаче ми - тогенного сигнала внутрь клетки. В составе димерного комплекса проис ходит взаимное трансфосфорилирование внутриклеточных составляющих молекул рецепторов. Подобное фосфорилирование рецепторов является важным механизмом регуляции ки - назной активности. Фосфорилированные аминокислоты рецепторов фор мируют зоны взаимодействия с после дующими компонентами цепи переда чи сигнала, которые осуществляются за счет упомянутых выше структурных, доменов.
Выявлено более 10 различных бел ков, носителей SН2-домена, способ ных связываться с фосфорилированными сайтами рецепторов P D G F . К числу таких белков относятся и неко торые ферменты: фосфоинозитол-3- киназа (ΡΙ3Κ), фосфолипаза С-γ (PLC-γ), киназы семейства src, тирозиновые фосфатазы и др. С рецепто рами P D G F ассоциируются и опреде
ленные адапторные гены: She, Nck, Crk и активаторы транскрипции (STAT). Известно, что соседствующие с фосфотирозином 3—6 аминокислот определяют специфичность связыва ния SН2-домена. Именно поэтому различные молекулы с SH2 взаимо действуют с определенным фосфорилированным сайтом белка-партнера.
Некоторые пути передачи сигнала, индуцированные β-рецептором PDGF и стимулирующие рост клетки и ее миграцию, схематично представлены на схеме 3.4.
Аномально повышенная активность P D G F или соответствующих рецепто ров может приводить к трансформации клетки. Это заключение впервые было сделано на основании открытия иден тичности В-цепи P D G F и вирусного онкогена sis. Позже было продемонст рировано, что суперэкспрессия PDGF или его рецептора характерна для не которых типов глиобластом и сарком человека. Злокачественное превраще ние P D G F и PDGF-рецептора проис-
116

С х е м а 3.4. Основные пути передачи сигналов, индуцированных Р-рецептором PDGF
src
РКС-белки
ходит в результате реализации всех описанных выше механизмов актива ции протоонкогенов:
• транслокация гена В-цепи P D G F с образованием химерных белков описана при дерматобластомах человека;
•формирование слитных белков, несущих р-рецептор P D G F с дру гими белками, обнаружено при остром миелоидном лейкозе;
•мутации в первичной структуре этих генов.
Имеется информация об актива ции рецептора P D G F в результате ин теграции в зону локализации этого ге на провируса HTLV1 при инфекции Т-клеток, т. е. описано уникальное явление инсерционного мутагенеза в клетках человека.
Естественно предположить, что об разование аберрантных форм этих белков нарушает нормальное функ ционирование систем передачи сигна лов с их участием.
117
3.1.3.2. Тирозинкиназы
всистеме передачи сигналов
Тирозинкиназы катализируют пе ренос фосфата от АТФ на тирозиновый остаток специфических клеточ ных белков-мишеней. Различные представители этой группы киназ во влечены в процессы, регулирующие клеточный рост и дифференцировку. Основные категории тирозинкиназ делятся на две группы: рецепторные и нерецепторные (см. табл. 3.4).
Все рецепторные тирозинкиназы имеют сходное строение — состоят из лигандсвязывающего внеклеточного участка, гидрофобного трансмембран ного домена и внутриклеточной (цитоплазматической) области. Послед няя наряду с каталитическим тирозинкиназным доменом содержит регуляторные зоны.
Нерецепторные тирозинкиназы яв ляются промежуточными проводника ми различных внутриклеточных сиг нальных путей. Многие из этих киназ ассоциированы с трансмембранными рецепторами, например рецепторами гормонов, цитокинами, рецепторами факторов роста и др. Активация нерецепторных киназ происходит в ре зультате ассоциации рецепторов с внеклеточными лигандами, или ком понентами клеточной адгезии, а также на определенных этапах клеточного цикла. Принципиальная схема систем передачи сигналов с участием рецепторных и нерецепторных тирозинки наз представлена на схеме 3.5.
Как и в случае стимуляции сигнала P D G F , ассоциация лиганда с рецепторными тирозинкиназами индуциру ет их димеризацию либо олигомеризацию. Эти конформационные измене ния ведут к индукции тирозинкиназной активности цитоплазматической области данного белка. Конформаци онные изменения белка играют ре шающую роль и в инициации нере цепторных тирозинкиназ.
Тонкие механизмы регуляции ти розинкиназ исследованы достаточно подробно. Рассмотрим молекулярные
механизмы регуляции тирозинкиназ на классическом примере онкобелка src. Молекула этого белка состоит из последовательно расположенных до менов: уникальный домен, специфич ный для каждого из представителей этого семейства белков, SНЗ-домен, SH2-домен, киназный домен и конце вой регуляторный элемент, несущий фосфорилированный тирозин (рТyr). В неактивной форме белка src рТyr связан с SН2-доменом, а SНЗ-домен — с начальной областью киназного до мена. Таким образом, за счет внутри молекулярных связей молекула белка принимает "закрытую" конформационную форму и не способна фосфорилировать субстратные белки. В нор ме фосфорилирование тирозина в регуляторном концевом фрагменте про изводится специфической киназой (Csk), а дефосфорилирование этого локуса — фосфатазами.
Альтернативным механизмом регу ляции активности src-белка и прохож дения сигнала является его специфи ческое взаимодействие с компонента ми системы передачи сигнала. В слу чае ассоциации SH2-домена с рТyr ка кого-либо другого белка или присое динения рТуг на конце src с SН2-до- меном белка-партнера киназная ак тивность инициируется. Ясно, что при нарушении мутаций в SH2- и SНЗ-до- менах, а также при утрате концевого Туг активность белка src становится нерегулируемой. Следует отметить, что основное отличие вирусного он кобелка v-src от клеточного аналога заключается именно в потере зоны, несущей регуляторный рТyr. Недавно было показано, что в метастазах рака толстой кишки человека белок c-src имеет нарушения в этой же области.
Активация как рецепторных, так и нерецепторных тирозинкиназ индуци рует каскад различных процессов фосфорилирования, которые включа ют аутофосфорилирование, фосфори лирование клеточных субстратов и во влечение SH2-доменсодержащих бел ков. В результате происходит последо вательная активизация ряда белков и
118
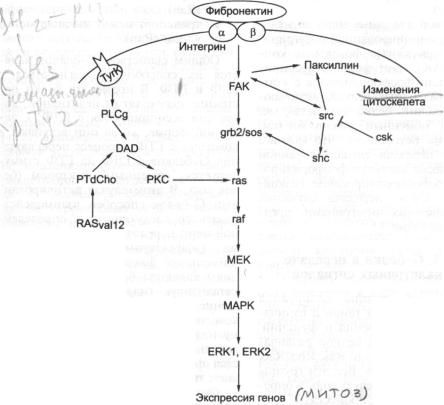
С х е м а 3.5. Основные пути передачи сигналов, стимулирован
ных интегринами
вторичных проводников сигнала, ко торые обладают специфичными регуляторными функциями. Эти сигналь ные импульсы достигают клеточного ядра и могут индуцировать экспрес сию определенных генов. Одним из наиболее исследованных сигнальных путей, где ключевую роль играет тирозинкиназа, являются цепи передачи сигналов от интегринов.
Интегрины представляют собой большой класс трансмембранных кле точных рецепторов, определяющих связь с внеклеточным матриксом (ЕСМ) и адгезивное взаимодействие между клетками, вследствие чего эти структуры вовлекаются во многие процессы в клетке — развитие эм бриона, апоптоз, реакция на стресс и пр. Специфичность взаимодействия
л
интегринов с определенными лигандами и внутриклеточными структура ми определяется сочетанием более 15 вариантов alpha- и beta-субъединиц, в ре зультате чего формируются рецепторы (в настоящее время известно более 20). Основными лигандами ЕСМ, ас социирующимися с интегринами, яв ляются фибронектин, коллаген и витронектин. Некоторые из интегринов способны связываться и с раствори мыми лигандами (например, фибри ноген). По аналогии с описанными выше механизмами активации рецеп торами факторов роста лиганды сти мулируют формирование кластеров интегринов, что ведет к образованию фокальных контактов и запуску соот ветствующих сигналов внутрь клетки. Многие из сигнальных белков, регу-
119
—г
лируемых интегринами, участвуют в процессах передачи сигналов, ини циированных другими рецепторами (см. выше).
Ключевой для стимуляции сигналь ной цепи, инициированной интегрина ми, является киназа фокальных кон тактов (FAK). Этот фермент ассоции руется и/или фосфорилируегя с ком плексом различных белков, включаю щим паксилин, белки семейства src, адаптеры, "обменный" фактор sos (см. ниже) и др. Естественно, что важным фактором передачи сигнала в данной системе также являются фосфорилирование и дефосфорилирование белковмишеней. Схема передачи сигналов, стимулированных интегринами, пред ставлена на схеме 3.5.
3.1.3.3. G-белки в передаче внутриклеточных сигналов
Белки этой группы кодируются большим семейством генов и отлича ются сходством строения и функций. Внутри семейства G-белков различа ют 5 крупных подгрупп: Ras, Rho/Rac, Rab, Ran и Rad/Gem. Все эти группы белков играют ключевую роль в опре деленных клеточных процессах:
•регуляция структуры цитоскелета (Rho/Rac);
•внутриклеточное движение (Rab);
•транспорт между цитоплазмой и ядром (Ran).
Общим свойством G-белков явля ется их способность связываться с ГДФ и ГТФ. В норме эти белки по стоянно переходят от неактивной, ко гда они ассоциируются с ГДФ, к ак тивной форме, когда они вступают в комплекс с ГТФ. Процесс переключе ния G-белков с ГДФ на ГТФ стиму лируется "обменным" фактором (бе лок sos). В этом случае активирован ный G-белок способен взаимодейст вовать со следующими в определен ной цепи передачи сигнала молекула ми (эффекторами). Антагонистами "обменного" фактора по своим функ циям являются'белки GAP, которые, катализируя гидролиз ГТФ (превра щение в ГДФ) в комплексе с G-бел- ком, переводят его в пассивную фор му (схема 3.6). Следует отметить, что активность GAP-белков контролиру ется фосфорилированием под дейст вием тирозинкиназ различных типов.
Фактически малые G-белки, на пример ras, являются распределителя
•пролиферация и дифференцими различных сигналов в клетке. Эти
ровка (Ras, Rho/Rac); |
белки интегрируют получаемые от |
"вышестоящих" |
молекул |
(компонен |
кулярных механизмов, в первую оче |
|||||||||||||||||||||
тов различных сигнальных путей) им |
редь вследствие мутаций в первичной |
|||||||||||||||||||||||
пульсы |
и |
активизируют последующие |
структуре (см. выше). |
|
|
|
|
|
||||||||||||||||
эффекторные белки, каждый из кото |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
рых |
представляет |
собой |
начальное |
3.1.3.4. "Ядерные" |
|
|
|
|
||||||||||||||||
звено следующего этапа определенной |
протоонкогены и факторы |
|
||||||||||||||||||||||
цепи передачи сигнала (см. схему 3.6). |
транскрипции в системе |
|
|
|||||||||||||||||||||
Сигнальные |
пути, |
приводящие к |
передачи сигналов |
|
|
|
|
|||||||||||||||||
активации белков ras, достаточно под |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
робно исследованы. В общем виде |
Многие из известных путей систе |
|||||||||||||||||||||||
они имеют сходное строение: внекле |
мы передачи сигналов в клетках име |
|||||||||||||||||||||||
точный |
лиганд |
(например, |
фактор |
ют своей целью индукцию определен |
||||||||||||||||||||
роста) присоединяется к экстрацеллю- |
ных клеточных генов, продукты экс |
|||||||||||||||||||||||
лярной части рецептора и стимулиру |
прессии которых необходимы клетке в |
|||||||||||||||||||||||
ет тирозинкиназную |
активность внут |
данный момент. Непосредственно ре |
||||||||||||||||||||||
риклеточного |
компонента |
этого |
бел |
гуляция |
этих |
|
генов |
|
осуществляется |
|||||||||||||||
ка. |
В |
результате |
фосфорилируются |
белками конечных звеньев цепей пе |
||||||||||||||||||||
другие тирозиновые остатки той же |
редачи |
сигналов |
- |
факторами транс |
||||||||||||||||||||
молекулы |
или |
|
промежуточные |
|
нере |
крипции, |
локализованными |
в |
ядре |
|||||||||||||||
цепторные киназы. С рТуг этих моле |
клетки. |
Большинство |
представителей |
|||||||||||||||||||||
кул за счет SH2-доменов взаимодейст |
данной |
категории |
|
генов |
являются |
|||||||||||||||||||
вуют последующие белки, которые ас |
протоонкогенами. |
|
|
|
|
|
|
|
||||||||||||||||
социируются |
с |
адапторами |
(Grb2, |
"Ядерные" |
|
протоонкогены |
|
могут |
||||||||||||||||
Shc). Последние в свою очередь инду |
быть отнесены к двум основным ти |
|||||||||||||||||||||||
цируют "обменный" фактор sos путем |
пам: |
гены, |
продуцирующие |
белки, |
||||||||||||||||||||
ассоциации своих SНЗ-доменов с |
способные |
взаимодействовать |
с |
кле |
||||||||||||||||||||
пролинбогатым |
участком |
белка |
sos. |
точной |
Д Н К |
только |
в |
виде |
комплек |
|||||||||||||||
Запуск |
"обменного" |
|
фактора активи |
сов |
с |
другими |
|
белками |
(например, |
|||||||||||||||
зирует белок ras, который через эф |
Fos-Jun, Мус и, возможно, Rel), и ге |
|||||||||||||||||||||||
фекторные |
белки |
распределяет |
им |
ны, |
кодирующие |
белки, |
специфично |
|||||||||||||||||
пульсы по отдельным сигнальным пу |
связывающиеся с определенными по |
|||||||||||||||||||||||
тям. |
|
|
|
|
|
|
|
|
|
|
следовательностями |
|
генома |
в |
моно |
|||||||||
Наиболее хорошо изученными эф- |
|
мерной форме (Ets, Myb, ErbA). Ак |
||||||||||||||||||||||
фекторами ras-белка являются Raf — |
тивность обеих групп факторов транс |
|||||||||||||||||||||||
стимулирует каскад МАР-киназ и |
крипции |
регулируется |
|
механизмами |
||||||||||||||||||||
фосфоинозитол-3-киназа (PI3K) — |
фосфорилирования. |
|
|
|
|
|
|
|
||||||||||||||||
активирует киназу Akt, что в конеч |
Белки семейств Fos и Jun являют |
|||||||||||||||||||||||
ном итоге предохраняет клетку от апоптоза. ся основными |
|
компонентами |
ком |
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
плексных |
факторов |
транскрипции |
|||||||||||
Нахождение G-белков на перекре |
API . Эти гетеродимеры функциони |
|||||||||||||||||||||||
стке различных путей передачи кле |
руют как позитивные либо негатив |
|||||||||||||||||||||||
точных сигналов определяет их важ |
ные |
факторы |
транскрипции |
|
генов |
|||||||||||||||||||
ную роль в реализации многих кле |
путем связывания |
с |
определенными |
|||||||||||||||||||||
точных процессов. Нарушение функ |
последовательностями |
Д Н К . |
Харак |
|||||||||||||||||||||
ций этих белков может привести к |
терной |
особенностью |
|
гетеродимер- |
||||||||||||||||||||
фатальным |
последствиям |
для |
|
орга |
ных |
факторов |
транскрипции |
являет |
||||||||||||||||
низма, в частности к трансформации |
ся существование в зоне специфиче |
|||||||||||||||||||||||
клетки. Этим объясняется относитель |
ского взаимодействия с Д Н К опреде |
|||||||||||||||||||||||
но высокая частота нарушений, на |
ленных |
последовательностей |
амино |
|||||||||||||||||||||
пример, протоонкогена Ras в различ |
кислот: 5 лейцинов, разделенных 6 |
|||||||||||||||||||||||
ных опухолях. Превращение протоон |
другими |
остатками |
|
(лейциновая |
||||||||||||||||||||
когенов ras в онкогены происходит в |
"змейка"), и две зоны alpha-спирали бел |
|||||||||||||||||||||||
результате |
действия |
различных |
моле- |
ка, |
разделенные |
участком |
beta-петли |
|||||||||||||||||
121
(структура спираль—петля—спираль). Эти структуры участвуют в формиро вании димерных комплексов и стаби лизируют взаимодействие белков с Д Н К . Белки семейства myc образуют ДНК-связывающие комплексы с бел ками группы МАХ.
Идентифицировано большое число клеточных генов, активность которых зависит от комплексных факторов транскрипции, кодируемых ядерными протоонкогенами. В норме эти факто ры участвуют в процессах пролифера ции, дифференцировки и в осуществ лении клеточного цикла.
Мономерные факторы транскрип ции содержат участки связывания с Д Н К , домен активации транскрипции и домен негативной регуляции. Каж дый из этих факторов узнает консенсусную последовательность в двуспиральной Д Н К клетки. Факторы транс крипции способны становиться онко генами в результате нарушений в структуре. Наиболее характерным ме ханизмом активации этих протоонко генов является утрата С-терминаль- ной области гена. Это изменение не затрагивает способности белков ука занных генов взаимодействовать с Д Н К , но нарушаются их функции ак тиваторов транскрипции. Именно та кие изменения в структуре вирусных онкогенов v-fos, v-erbA и v-myb отли чают их от соответствующих клеточ ных онкогенов.
3.1.3.5. Протоонкогены — регуляторы гибели клетки
С открытием гена BCL2, функция которого заключается в негативной регуляции программированной гибе ли клеток (апоптоза), стало ясно, что онкогенным потенциалом могут об ладать не только гены, вовлеченные в механизмы клеточной пролифера ции. Гены семейства BCL2 являются компонентами внутриклеточных "сверочных точек" апоптотических сигналов, индуцированных различ ными факторами. В норме гибель клеток представляет собой строго ре-
гулируемый процесс. Система кон троля апоптоза с участием генов се мейства BCL2 построена по следую щей схеме. Под действием различных экзогенных факторов (оксидативный стресс, облучение, утрата цитокинов, генотоксические повреждения) про исходит стимуляция "импульсов смерти", которые запускают систему программированной гибели клеток и через ряд промежуточных этапов первой "сверочной точки" апоптоза. В комплекс "сверочной точки" входят
локализованные |
на |
митохондриях |
|||
белки с |
противоположными |
свойст |
|||
вами: |
индукторы |
апоптоза |
(гены |
||
ВАХ, ВАК, BAD и др.) и соответст |
|||||
вующие |
антагонисты |
— |
негативные |
||
регуляторы апоптоза |
(гены |
BCL2, |
|||
BCL-LX, BCL-W |
и др.). |
Соотноше |
|||
ние индукторов и негативных регуля торов апоптоза определяет реакцию клетки на сигналы гибели. Группа белков с противоположными функ циями имеет сходные консерватив ные домены (ВН1, ВН2, ВНЗ, ВН4, ТМ), которые осуществляют физиче ское взаимодействие этих белков — образование активных димерных структур и дальнейшую передачу сиг нала. На следующем этапе апоптоза (фаза экзекуции) происходит индук ция белков семейства каспаз, кото рые представляют собой цистеиновые протеазы, специфически расще пляющие субстраты по остаткам аспарагиновой кислоты. Активирован ные каспазы формируют генеротетрамерные комплексы, разрушающие эндогенные белки-мишени (PARK в ядре, L4-gDl в цитоплазме). После прохождения данной стадии процесс апоптоза становится необратимым и клетки гибнут.
Описанная система внутриклеточ ных молекулярных механизмов, во влеченных в апоптоз, весьма консер вативна и описана как у позвоночных, так и у беспозвоночных. Нарушение этих процессов, т. е. потеря клетками способности к гибели, может приво дить к стимуляции опухолеродного потенциала клетки.
122
Как отмечалось в начале данного раздела, нормальное функционирова ние системы передачи сигналов в клетке обеспечивает ее полноценное существование в окружающей среде и взаимодействие с другими клетками. При малигнизации наблюдается об ратная ситуация — сигнальная систе ма перестраивается таким образом, чтобы обеспечить трансформирован ной клетке независимость от окру жающей среды и тканей. Фактически трансформация — это аберрантная версия системы передачи сигналов в клетке, поэтому в раковой клетке в подавляющем большинстве случаев происходит изменение нескольких белков, вовлеченных в систему пере дачи сигнала.
Вполне естественно, что изменен ные белки должны относиться к раз личным сигнальным цепям и нахо диться на различных этапах прохож дения сигнала в клетке. Этот вывод имеет многочисленные эксперимен тальные подтверждения. В частности, об этом свидетельствует тот факт, что большинство известных онкогенов от носится к ключевым белкам сигналь ной системы клетки — факторам рос та, трансмембранным рецепторам, внутриклеточным эффекторам, фак торам транскрипции и т. д.
С современной точки зрения рак является генетическим заболевани ем (заболеванием генома клетки!), вызванным изменениями в протоонкогенах (или генах-супрессорах) и выражающимся на молекуляр ном уровне нарушением системы передачи сигналов в клетке.
** *
Взаключение необходимо отме тить, что концепция онкогена, т. е. дискретного носителя генетической информации, который при опреде ленных условиях способен вызывать трансформацию нормальной клетки, впервые в истории онкологии позво лила объединить основные направле
ния исследований канцерогенеза. Практически все канцерогенные фак торы — химические, физические, на следственные и т. д. — приводят к по вреждению онкогенных генетических детерминант (протоонкогенов, геновсупрессоров) и их функций.
Нарушения в структуре генов мо гут происходить без видимых воздей ствий Каждую секунду в организме человека происходит деление около 25 млн клеток. Этот процесс осущест вляется под строгим контролем, в оп ределенных органах и в точное время. Контроль производится комплексом молекулярных систем, механизмы функционирования которых, к сожа лению, еще не установлены. Подсчи тано, что каждый ген (их примерно 50 тыс. в клетке человека) в процессе жизнедеятельности организма подвер гается спонтанным нарушениям око ло 1 млн раз. Все нарушения фикси руются и устраняются системами ре парации клеточного генома. В редчай ших случаях нормальная структура из мененного гена не восстанавливается, кодируемый белковый продукт и его свойства изменяются, и если эта ано малия имеет принципиальный харак тер и затрагивает ключевые гены (по тенциальные онкогены), становится возможной трансформация клетки. Надо полагать, что, за редким исклю чением (например, при вирусиндуцированном канцерогенезе), для подоб ных событий недостаточно 1—2 мута ций или перестроек в геноме, однако за годы жизнедеятельности наруше ния в геноме накапливаются и могут повлечь за собой качественные изме нения в молекулярных механизмах ре гуляции основных процессов, проис ходящих в клетке, что ведет к индук ции опухолевого процесса.
Повреждения, затрагивающие по тенциальные онкогены, могут быть двух типов:
• в половых клетках. В этом случае они наследуются и можно гово рить о наследуемых семейных формах рака;
123
•в соматических клетках. В этом факторов (признаков). Расшифровка
случае они не наследуются, но оп ределяют трансформацию именно той клетки, которая их приобре тает. Большинство известных ра ков относится к данному типу.
На оснований представленной в этой главе информации можно сфор мулировать несколько гипотетических положений о принципах взаимодейст вия онкогенных детерминант в про цессе злокачественной трансформа ции клеток. Существует оптимум (предел) экспрессии онкогенных де терминант, приводящих к злокачест венной трансформации клетки. Эф фект может быть достигнут как в ре зультате действия одного сильного фактора, так и вследствие накопления нескольких менее сильных факторов.
Клетки не "терпят" экспрессии двух и более онкогенов, каждый из которых способен успешно их транс формировать: выбирается лишь один — активность остальных подавляется. При "параллельной" избыточной экс прессии подобных генов клетка гиб нет. Для "защиты" от гибели исполь зуются различные молекулярные ме ханизмы: метилирование генов, коди рующих избыточные факторы транс формации, делеция этих факторов, деградация соответствующих белков, расщепление информационной Р Н К в результате действия механизма Р Н К - интерференции (RNAi) и др.
В трансформации клеток, индуци
рованной |
определенным |
фактором |
||
(онкогеном), |
не участвуют |
факторы |
||
сходного |
типа |
(представители |
одного |
|
и того же семейства генов). |
|
|
||
Малигнизированная клетка |
стре |
|||
мится сохранить "статус кво", т. е. за щищается и от "лишних" малигнизирующих факторов, и от факторов, ве дущих к нормальному фенотипу.
Все вышеупомянутые гипотетиче ские механизмы взаимодействия он когенных детерминант реализуются в результате селекции клеток с соответ ствующим сочетанием необходимых
основных молекулярных механизмов и принципов индукции и развития опухолевого процесса, которая несо мненно связана с поразительными ус пехами молекулярной онкологии, по зволяет сделать также и ряд общих предположений, имеющих практиче ское значение.
Воснове различных онкологиче ских заболеваний лежат сходные мо лекулярные процессы, т. е. вовлечены родственные группы генов и белков. Среди потенциальных онкогенов име ется ограниченное число ключевых генов, аномалии в которых должны встречаться в большинстве неоплазий.
Виндукции и развитии неоплазий одного типа могут участвовать различ ные опухолевые гены, но при этом должны существовать определенные гены, нарушения которых обязательно для данной формы рака.
Вышеизложенное дает надежду, что бурно развивающиеся в последние годы методы генотерапии и сигналотерапии позволят в обозримом буду щем на принципиально новом уровне решать вопросы лечения и предотвра щения онкологических болезней.
Рекомендуемая литература
Hesketh R. The Oncogene Handbook Aca demic press. — New York, 1995.
Lodish H. et al. Molecular Cell Biology // Scient. Amer. Books, Inc. — New York, 1995.
Brandel C, Tooze J. Introduction to Protein Structure. — Garland Publishing, Inc. — New York, 1998.
Robinson D., Wu Y. M., Lin S. F. The protein tyrosine kinase family of the human ge nome // Oncogene. — 2000. - Vol. 19. - P. 5548-5557.
Hunter T. Signaling-2000 and Beyond // Cell. - 2000, January, 7. - Vol. 100. - P. 113-127.
Nigg E. Mitotic kinases as regulators of cell di vision and its checkpoints // Nature Rev.
-2001. - Vol. 2. - P. 21 - 32 .
Klausner R. The fabric of cancer cell biologyweaving together the strands // Cancer Cell. - 2002. - Vol. 1. - P. 3-10.
124

3.2. Опухолевые супрессоры |
собом |
увеличивает темп |
возникнове |
|||||||||||||||
и мутаторные гены |
|
|
|
ния мутаций и/или других генетиче |
||||||||||||||
Б. П. Копнин |
|
|
|
|
|
|
|
ских изменений. Следует заметить, |
||||||||||
|
|
|
|
|
|
|
что многие из опухолевых супрессо |
|||||||||||
Ключевую |
роль |
в |
возникновении |
ров являются одновременно и мута- |
||||||||||||||
торными генами. Инактивация таких |
||||||||||||||||||
важнейших |
свойств |
неопластической |
||||||||||||||||
генов столь сильно увеличивает веро |
||||||||||||||||||
клетки |
|
(см. |
главу 2) |
играют |
наруше |
|||||||||||||
|
ятность появления различных онко- |
|||||||||||||||||
ния |
функции |
протоонкогенов |
(см. |
|||||||||||||||
генных мутаций, что образование опу |
||||||||||||||||||
раздел |
3.1), |
опухолевых супрессоров |
и |
|||||||||||||||
холи становится лишь делом времени. |
||||||||||||||||||
так |
называемых |
мутаторных |
генов. |
|||||||||||||||
Действительно, врожденные мутации |
||||||||||||||||||
Термином |
опухолевые |
супрессоры |
(дру |
|||||||||||||||
даже в одном из аллелей некоторых из |
||||||||||||||||||
гие |
названия |
— |
антионкогены, |
рецес |
||||||||||||||
опухолевых |
супрессоров/мутаторных |
|||||||||||||||||
сивные |
опухолевые |
гены) |
принято |
обо |
||||||||||||||
генов |
делают |
вероятным |
возникнове |
|||||||||||||||
значать |
гены, |
инактивация |
функции |
|||||||||||||||
ние в молодом возрасте определенных |
||||||||||||||||||
которых |
ведет |
к |
возникновению |
и/ |
||||||||||||||
форм |
новообразований |
- |
сарком, |
|||||||||||||||
или |
|
профессии |
|
новообразований. |
||||||||||||||
|
|
лейкозов, опухолей мозга или рака |
||||||||||||||||
Восстановление же |
экспрессии |
неко |
||||||||||||||||
молочной железы (синдром Ли — |
||||||||||||||||||
торых |
из таких |
генов |
в опухолевых |
|||||||||||||||
Фраумени) — при мутациях р53; рака |
||||||||||||||||||
клетках, |
наоборот, может подавить их |
|||||||||||||||||
молочной железы и яичников — при |
||||||||||||||||||
дальнейшее |
размножение. |
Мутатор- |
||||||||||||||||
мутациях генов BRCA1 и BRCA2 и т. д. |
||||||||||||||||||
ными |
|
называют |
гены, |
нарушение |
||||||||||||||
|
(табл. |
3.5). |
|
|
|
|||||||||||||
функции которых тем или иным спо |
|
|
|
|||||||||||||||
|
|
|
|
|
||||||||||||||
Т а б л и ц а |
3.5. Основные |
характеристики |
некоторых идентифицированных |
опухолевых |
||||||||||||||
супрессоров и мутаторных генов |
|
|
|
|
|
|
|
|
||||||||||
|
Хромо |
|
Ген |
сомная |
|
локали |
||
|
||
|
зация |
|
р53 |
17р13 |
|
ΙΝΚ4a * |
9р21 |
|
Rb |
13ql4 |
|
TβR-II |
3р22 |
|
SMAD2, |
18q21 |
|
SMAD3 |
15q21-22 |
SMAD4/DPC4
18q21
CDH1 (Е-кад-
герин) 16q24
Новообразования* |
Функция белка |
Синдром Ли—Фраумени |
Регуляция клеточного цикла, |
(раздел 3.2.2.1) и большинст |
апоптоза, репарации Д Н К ; |
во форм спорадических опу |
поддержание целости генома |
холей |
|
Наследственная меланома и |
Ингибирование Cdk4 |
многие спорадические опу |
(pl6INK4a), активация р53 |
холи |
(р14ARF) |
Наследственная ретинобла- |
Контролирует вход в S-фазу и |
стома и остеосаркома; многие |
дифференцировку клеток |
формы спорадических опухо |
|
лей |
|
Наследственный и споради |
Рецептор второго типа для |
ческий рак толстой кишки |
T G F - β |
Рак толстой кишки, легкого, |
Передают сигнал от активиро |
поджелудочной железы |
ванных рецепторов T G F - β к |
|
Smad4 |
Ювенильный гамартоматоз- |
Транскрипционный фактор, |
ный полипоз желудка и ки |
опосредует действие T G F - β |
шечника; различные формы |
|
спорадических опухолей |
|
Наследственный рак желудка |
Участвует в межклеточных |
и многие формы спорадиче |
взаимодействиях, регулирует |
ской опухоли |
активность β-катенина |
125

|
|
Хромо |
|
Ген |
сомная |
|
локали |
|
|
|
|
|
|
зация |
АРС |
|
5q21 |
ΑΧΙΝ |
|
16р13.3 |
VHL |
|
3р25-26 |
WT1 |
|
11р13 |
PTEN/MMAC1 |
10q23.3 |
|
NF1 (нейро- |
17ql 1.2 |
|
фибромин) |
|
|
NF2 (мерлин) |
22ql 1.1 |
|
ATM |
|
11q23.1 |
NBS1 |
|
8q21 |
СНК2 |
|
22q |
BRCA1 |
|
17q21 |
BRCA2 |
|
13ql2 |
USH2, |
MSH6, |
2p-16 |
MLH1, |
PMS2 |
2pl5-16 |
|
|
3p21.3 |
|
|
7p22 |
|
Продолжение |
|
Новообразования* |
Функция белка |
|
Наследственный аденоматоз- |
Регулирует стабильность и |
|
ный полипоз и спорадиче |
транскрипционную актив |
|
ские опухоли толстой кишки |
ность β-катенина |
|
Гепатоцеллюлярный рак, |
То же |
|
опухоли толстой кишки |
|
|
Синдром фон Хиппеля—Лин- |
Подавляет экспрессию гена |
|
дау (раздел 3.2.7); светлокле- |
V E G F (фактора роста эндоте |
|
точная карцинома почки |
лия сосудов) и других генов, |
|
|
активируемых при гипоксии |
|
Наследственная нефробласто- |
Регуляция урогенитальной |
|
ма (опухоль Вильмса) |
дифференцировки |
|
Болезнь Коудена (раздел |
Стимуляция апоптоза, инги- |
|
3.2.4), многие спорадические |
бирование входа в S-фазу кле |
|
опухоли |
точного цикла |
|
Нейрофиброматоз 1-го типа |
Переводит гены ras из актив |
|
|
ной в неактивную форму |
|
Нейрофиброматоз 2-го типа; |
Осуществляет связь мембраны |
|
с цитоскелетом, обеспечивает |
||
менингиома, мезотелиома и |
||
другие опухоли |
контактное торможение деле |
|
|
ния |
|
Атаксия-телеангйэктазия |
При повреждениях Д Н К акти |
|
(раздел 3.2.10.1), спорадиче |
вирует р53, NBS1, BRCA1/2 |
|
ские лимфолейкозы |
и др. |
|
Ниймегенский синдром (раз |
При повреждениях Д Н К акти |
|
дел 3.2.10.1), спорадические |
вирует СНК2, BRCA1 и др. |
|
лимфолейкозы |
|
|
Вариант синдрома Ли—Фрау- |
При повреждениях Д Н К акти |
|
|
вирует р53 и BRCA1 |
|
Наследственные опухоли мо |
Модулирует активность мно |
|
лочной железы и яичников |
гих факторов транскрипции, |
|
|
участвует в репарации ДНК |
|
То же |
Модулирует транскрипцию ге |
|
|
нов, участвует в репарации |
|
|
Д Н К |
|
Неполипозный рак толстой |
Репарация неспаренных уча |
|
кишки и яичников; многие |
стков Д Н К (mismatch repair) |
|
спорадические опухоли |
|
* Подчеркнуты наследственные формы заболеваний, возникающие при мутациях в половых
клетках.
**Ген INK4a кодирует два белка: p16INK4a и p14ARF (Alternative Reading Frame) — продукт аль тернативной рамки считывания. Делеции и многие точечные мутации в гене 1ΝΚ4a вызывают одновременную инактивацию супрессорных активностей обоих этих белков.
Доказательством причинной роли таких мутаций в канцерогенезе явля ются результаты экспериментов по созданию линий трансгенных мышей,
во всех клетках которых инактивированы ("нокаутированы") один или оба аллеля какого-либо из опухолевых су прессоров или мутаторных генов. Если
126
такие изменения не вызывают внутри утробную гибель, то у рождающихся животных возникает сильная предрас положенность к развитию определен ных новообразований. Так, мыши с инактивированным р53 характеризуют ся почти 100 % вероятностью развития в молодом возрасте новообразований, спектр которых сходен с наблюдаемым при синдроме Ли—Фраумени.
Геном человека содержит по мень шей мере несколько десятков опухо левых супрессоров и мутаторных ге нов. Более 30 из них уже идентифици рованы, для многих известны выпол няемые в клетке функции (см. табл. 3.5). Выявлено еще несколько десят ков участков хромосом, потери кото рых обнаруживаются во многих случа ях какой-то определенной формы опухолей или в различных новообра зованиях, что указывает на возмож ную локализацию в них потенциаль ных опухолевых супрессоров. Ниже будут рассмотрены основные сведения о функциях известных опухолевых супрессоров/мутаторных генах и воз можных механизмах возникновения новообразований при их нарушениях.
3.2.1. pRb— первый идентифицированный опухолевый супрессор
3.2.1.1. Открытие гена Rb
Идентификация опухолевых су прессоров началась с обнаружения ге на Rb, врожденные мутации которого вызывают развитие ретинобластом. В начале 70-х годов Кнудсон, проводя эпидемиологические исследования, от метил, что около 40 % ретинобластом возникает в младенческом возрасте (средний возраст 14 мес), причем эти опухоли, как правило, билатеральные (возникают из сетчатки обоих глаз) и часто множественные (в среднем по 3 независимых опухоли на пациента). Если такие пациенты в результате хи рургического вмешательства излечива лись от ретинобластом, у многих из них в юношеском возрасте развивалась
остеосаркома, а в зрелом возрасте — меланома кожи. При этом во многих случаях характер заболевания был на следственным. Кнудсон сформулиро вал следующую гипотезу: если дети на следуют мутантный аллель гена, позже названного Rb, то вторая мутация, про исходящая уже в ретинобласте, ведет к возникновению опухоли. Это наблюда ется довольно часто (примерно у 95 % пациентов с частотой около трех неза висимых опухолей на пациента), по этому предрасположение к развитию болезни имеет доминантный характер наследования. Кнудсон предположил, что дети, у которых ретинобластомы возникают в более позднем возрасте (частота таких опухолей невелика — 1 на 30 000 детей), не наследуют мутант ный аллель гена Rb. Вместо этого у них происходят две независимые мутации в одном из ретинобластов, что и приво дит к развитию опухоли. Поэтому, со гласно гипотезе Кнудсона, у пациентов первой группы имеется одна врожден ная и одна приобретенная мутации, то гда как у больных второй группы обе мутации приобретенные.
В связи с тем что при некоторых наследственных ретинобластомах обна руживались небольшие делеции участ ка длинного плеча хромосомы 13 (13ql4), было предположено, что ген "предрасположенности к ретинобластоме" (Rb) локализуется именно в этом участке генома. И действительно, с помощью позиционного клонирова ния был изолирован ген, оба аллеля которого инактивированы в клетках как наследственных, так и спорадиче ских ретинобластом. При этом при на следственных формах заболевания все клетки организма имели врожденные мутации этого гена. Таким образом, стало ясно, что постулируемые Кнудсоном две мутации, необходимые для развития ретинобластом, происходят в разных аллелях одного и того же гена Rb. Инактивация гена Rb была обнару жена не только в ретинобластомах, но и в клетках некоторых других, нена следственных новообразований: прак тически во всех случаях мелкоклеточ-
127
ного рака легкого, части случаев (20— 40 %) острого лейкоза, остеосаркомы, рака мочевого пузыря, предстательной железы и др. Восстановление экспрес сии этого гена в культивируемых in vit ro клетках ретинобластом и остеосарком приводило к торможению их рос та. Таким образом, впервые были по лучены веские доказательства сущест вования генов, полная инактивация которых приводит к развитию новооб разований и продукты которых способ ны подавить размножение неопласти ческих клеток. Закономерности, выяв ленные при исследовании гена Rb, в частности ассоциация с наследствен ными формами опухолей и необходи мость поражения обоих аллелей (ре цессивный характер проявления мута ций), стали использоваться в качестве критериев при поиске и идентифика ции других опухолевых супрессоров.
Сейчас достигнут значительный прогресс в понимании нормальной функции продукта гена Rb (pRb) в клетке, охарактеризованы типы и ме стоположение мутаций в гене Rb при ретинобластомах и других новообра зованиях, предложены высокоэффек тивные методы скрининга мутаций Rb у индивидуумов в группах риска.
3.2.1.2.Функция pRb в клетке
иее нарушения
при канцерогенезе
pRb представляет собой фосфобелок с мол. массой 105 кДа, локализую щийся в ядре и экспрессирующийся в большинстве типов клеток. Он дефосфорилирован в неделящихся клетках, а также в пролиферирующих клетках, находящихся в начале Gl - фазы кле точного цикла. В таком состоянии pRb образует комплексы с рядом белков, в том числе с белками, вызывающими ремоделирование хроматина (гистоновые деацетилазы HDAC, комплексы SWI—SNF), и транскрипционными факторами семейства E2F, регулирую щими активность генов, продукты ко торых необходимы для начала и прохо ждения S-фазы (циклин Е, циклин А,
дигидрофолатредуктаза, тимидинкиназа, PCNA, ДНК-полимераза а и др). Транскрипция этих генов подавлена, если E2F связан с комплексами HDAC/pRb/SWI-SNF. При митогенных сигналах pRb в середине Gl-фазы фосфорилируется по определенным аминокислотным остаткам циклинзависимой киназой циклин D/cdk4(6) (см. раздел 2.2.1), что вызывает отвя зывание от него HDAC. Комплекс pRb/SWI - SN (без HDAC) не блокиру ет способность E2F трйнс-активиро- вать ген циклина Е, но репрессирует транскрипцию других E2F-peагирye- мых генов, в частности циклина А. В результате в конце Gl-фазы происхо дит избирательная активация циклинзависимой киназы циклин E/cdk2, ко торая дополнительно фосфорилирует pRb по другим аминокислотным остат кам. Это вызывает высвобождение транскрипционного фактора E2F из комплексов с pRb/SWI—SNF и его ак тивацию, приводящую к повышению экспрессии циклина А и других генов, продукты которых необходимы для синтеза Д Н К (схема 3.7).
После завершения S-фазы pRb пе реходит в дефосфорилированное со стояние, в котором он блокирует ак тивность E2F и вход в следующую S- фазу (для ее инициации необходим новый митогенный стимул, активи рующий комплексы циклин D/cdk4). Таким образом, модулируя активность E2F и регулируемых им генов, pRb играет ключевую роль в контроле по следовательности событий, обеспечи вающих переход клетки из G0/G1 в S- фазу и ее успешное завершение.
Вдополнение к E2F pRb связывает
имодулирует активность ряда других транскрипционных факторов, боль шинство из которых тканеспецифичны
иучаствуют в регуляции определенных дифференцировочных программ. Так, он увеличивает транскрипционную ак тивность некоторых представителей се мейства bHLH (MyoD и др.), которые стимулируют мышечную дифференцировку и семейства С/ЕВР, играющих ключевую роль в моноцитарно-макро-
128
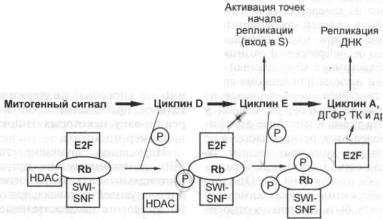
С х е м а 3.7. pRb регулирует активность транскрипционных фак
торов семейства E2F и цикл независимых киназ, ответственных за вход и продвижение по S-фазе клеточного цикла (объяснение
в тексте)
фагальной (NF-IL6) и адипоцитарной (С/ЕВР-c) дифференцировках. Кроме того, pRb связывает и репрессирует гомеобокссодержащие транскрипцион ные факторы (Рах-3, Mhox, Chx10), определяющие судьбу клетки в раннем эмбриогенезе. Все это дало основание полагать, что основная физиологиче ская функция pRb заключается в де терминировании некоторых терми нальных дифференцировок, для чего необходимы остановка клеточного цикла (осуществляется за счет подавле ния функции E2F) и индукция экс прессии ряда специализированных белков (реализуется путем модифика ции активности тканеспецифичных транскрипционных факторов).
Значительная часть мутаций гена Rb, обнаруживаемых в различных опу холях, вызывают либо делецию гена, либо сдвиг кодирующей рамки, либо ее преждевременную терминацию, ли бо нарушения сплайсинга м Р Н К . Все это приводит или к полной потере экспрессии белкового продукта, или к экспрессии неполноценных и неста бильных белков pRb. В части случаев наблюдаются миссенс-мутации, вызы вающие замену одного из аминокис лотных остатков. Такие мутации пора жают домен, ответственный за связы
вание pRb с рядом клеточных белков и вирусными онкобелками — T-SV40, Ε1 Α-аденовирусов и Е7 HPV (см. раз дел 5.2.9.1). По-видимому, нарушение функции именно этого домена, вызы ваемое либо мутациями, либо его свя зыванием с вирусными онкобелками, является критичным для подавления супрессорной активности pRb.
При мутациях обоих аллелей гена Rb, вызывающих отсутствие в клетке белка pRb или экспрессию его функ ционально-неактивной формы, транс крипционный фактор E2F находится в перманентно активированном состоя нии. Это, во-первых, уменьшает зави симость размножения клеток от росто вых факторов, а во-вторых, отменяет негативную регуляцию клеточной про лиферации при ростингибирующих сигналах (см. раздел 2.2.1). Кроме того, потеря функции pRb нарушает процес сы клеточной дифференцировки. Все это резко увеличивает вероятность по явления постоянно пролиферирующих клонов клеток, в которых будут накап ливаться и другие онкогенные мута ции, ведущие к злокачественной трансформации. При этом пока остает ся неясным, почему при врожденной инактивации одного из аллелей гена Rb во всех клетках организма у паци-
9-7908 Д. Г. Заридзе
ента в юном возрасте развивается именно ретинобластома, а не какое-то другое новообразование. Интересно, что у трансгенных мышей с инактива цией одного из аллелей гена Rb (инак тивация обоих аллелей несовместима с жизнью эмбрионов из-за нарушений эритропоэза и нейрогенеза) возникает не ретинобластома, а медуллярный рак щитовидной железы или аденома сред ней доли гипофиза, появляющаяся из меланофоров (в отличие от мышей у человека эта ткань в гипофизе атрофи рована). Более того, ретинобластома не развивается и у химерных мышей, во всех клетках сетчатки которых инактивированы оба аллеля гена Rb. Отсутст вие ретинобластомы у таких химерных мышей может быть объяснено относи тельно небольшим по сравнению с че ловеком количеством клеток в сетчат ке, что уменьшает вероятность возник новения в течение короткой жизни животного в одном из ретинобластов дополнительных генетических измене ний, необходимых для развития ново образования. Меньшая вероятность та ких событий может быть также связана с отсутствием у лабораторных мышей дополнительных канцерогенных фак торов, таких как облучение клеток сет чатки солнечным светом, что, очевид но, способствует развитию ретинобластом в аналогичной ситуации у челове ка. В пользу предположения о необхо димости для развития ретинобластом дополнительных событий может свиде тельствовать тот факт, что ретинобла стомы возникают у трансгенных мы шей, экспрессирующих вирусный онкобелок T-SV40 (см. раздел 5.2.8.2), ко торый связывает и инактивирует как pRb, так и его гомологи (см. ниже), а также другой опухолевый супрессор — р53 (см. раздел 3.2.2).
3.2.1.3. Гомологи pRb: р107 и Rb2/pl30
Несколько позже, в начале 90-х го дов, были идентифицированы два ге на, продукты которых, белки p107 и Rb2/p130, имеют структурное сходст
во и частично перекрывающиеся функции с pRb. Так, подобно pRb, они способны подавлять активность Е2F-респонсивных генов и блокиро вать вход в фазу S. Вместе с тем они имеют ряд отличий от pRb, в частно сти связывают только E2F4 и E2F5, тогда как pRb взаимодействует с E2F1, E2F2, E2F3 и E2F4. Возможно, поэтому р107 и Rb2/pl30 в отличие от pRb не способны поддерживать дли тельное пребывание в GO и дифференцировку некоторых типов клеток, например миоцитов.
Нарушения функции гомологов pRb, по-видимому, не причастны к инициальным этапам развития какихлибо опухолей человека, так как пока не выявлены наследственные формы новообразований с врожденными тер минальными мутациями генов p107 или Rb2/p130. Кроме того, у мышей с гетерозиготным нокаутом этих генов частота возникновения опухолей не повышается. В то же время соматиче ские инактивирующие мутации гена Rb2/p130 характерны для части случа ев лимфомы Беркитта, рака носоглот ки и мелкоклеточного рака легкого, причем, как правило, они выявляются на поздних стадиях заболевания. Ве роятно, нарушения функции этого го молога pRb обеспечивают прогрессию некоторых новообразований.
3.2.2. р53 — многофункциональный опухолевый супрессор, чаще всего поражаемый в различных
новообразованиях человека
3.2.2.1. Типы опухолей, ассоциированные с аномалиями р53
Наиболее универсальным молеку лярным изменением в различных но вообразованиях человека является инактивация функции белка р53. Ча ще чем в половине всех опухолей че ловека (50—60 % новообразований более чем 50 различных типов) обна-
130
руживаются мутации гена р53. В отли чие от других опухолевых супрессоров, для которых характерны мутации, прекращающие синтез белка (делеции, образование стоп-кодонов, сдвиг кодирующей рамки, нарушения сплай синга мРНК), подавляющее большин ство (более 90 %) мутаций р53 пред ставляют собой миссенс-мутации, при водящие к замене одной из аминокис лот в белковой молекуле на другую. Еще одной особенностью мутаций р53 в опухолевых клетках является то, что они в отличие от мутаций других опу холевых супрессоров часто гетерози готны, т. е. поражают один из двух ал лелей гена (причины этих различий будут раскрыты при рассмотрении структурной организации и функций р53 — см. раздел 3.2.2.2).
Мутации обнаруживаются в разных участках молекулы р53, но чаще всего в его эволюционно-консервативном ДНК-связывающем домене, причем с наибольшей частотой в кодонах 175, 245, 248, 249, 273 и 282 (так называе мые горячие точки) (см. рис. 3.8). Ин тересно, что спектр мутаций несколь ко меняется в зависимости от гистоге неза опухоли и/или этиологического фактора. Например, мутации в кодоне 175 не встречаются в опухолях легких, а замены в другой горячей точке — кодоне 273 — не обнаруживаются при бластном кризе хронического миелоидного лейкоза. В то же время для ра ка легкого характерны мутации в ко доне 145, очень редко встречающиеся
вдругих новообразованиях, а мутации
вкодоне 249 обнаруживаются преиму щественно в гепатокарциномах, вы званных специфическим канцероге ном — афлатоксином В. Очевидно, отражением действия канцерогенов с разными механизмами мутагенного действия являются и различия в ха рактере мутаций р53 в разных опухо лях. Так, при опухолях легких, печени и лимфомах замена аминокислотных остатков в большинстве случаев обу словлена трансверсиями (в Д Н К пуриновый нуклеотид заменен пиримидиновым или наоборот), а при карци
номах кожи, лимфоме Беркитта, Т- клеточном лейкозе — транзициями (заменой пуринового основания дру гим пуриновым или пиримидинового другим пиримидиновым).
Терминальные (произошедшие в половой клетке и передающиеся по на следству) мутации в одном из аллелей гена р53 вызывают синдром Ли—Фрау- мени, заключающийся во врожденном предрасположении к развитию различ ных новообразований, в первую оче редь сарком, рака молочной железы, лимфолейкозов. Нередко синдром Ли—Фраумени характеризуется воз никновением первично-множествен ных опухолей. Примечательно, что у трансгенных мышей, несущих инактивирующие мутации в гене р53, наблю дается картина, очень напоминающая синдром Ли—Фраумени. Примерно у 1/3 животных, у которых инактивирован один из двух аллелей р53, в тече ние 6—9 мес после рождения возника ют новообразования, причем их спектр очень сходен с наблюдаемым при син дроме Ли—Фраумени. При этом в час ти этих опухолей, как и новообразова ний у пациентов с синдромом Ли — Фраумени, сохраняется экспрессия не поврежденного аллеля гена р53. При врожденной инактивации во всех клет ках организма обоих аллелей гена р53 опухоли развиваются практически у всех животных. Примерно такая же картина наблюдается у трансгенных мышей, несущих дополнительный эк зогенный аллель р53, кодирующий бе лок с миссенс-мутацией.
Важно подчеркнуть, что мутации — не единственный путь нарушения функции белка р53 в опухолевых клет ках. Так, для 10—20 % рака молочной железы, а также для нейробластом ха рактерно нарушение транспорта р53 из цитоплазмы в ядро, где он проявляет свою функциональную активность. В части остеосарком наблюдается ампли фикация клеточного онкогена MDM2, продукт которого связывает и инактивирует белок р53. При раке шейки мат ки, ассоциированном с вирусами па пиллом человека, происходит связыва-
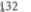
ние р53 с вирусным онкобелком Е6 (см. раздел 5.3.3), что вызывает дегра дацию белка р53, и т. д.
3.2.2.2. Структурная организация и биохимические активности белка р53
Продукт гена p53 имеет мол. массу 53 кДа и состоит из 392 аминокислот ных остатков. Он образует тетрамерный комплекс, способный регулиро вать транскрипцию ряда генов, имею щих в своем составе специфические последовательности Д Н К , так назы ваемые р53-респонсивные элементы. В молекуле р53 картировано несколь ко функционально-значимых доме нов, играющих важную роль в осуще ствлении или регуляции его активно сти (схема 3.8).
N-концевой участок (аминокисло ты 1—42) представляет собой домен, ответственный за транскрипционную активацию генов-мишеней. Он обла дает способностью связываться с ком понентами базальных факторов транс крипции, в частности с субъединица ми hTAFII31, hTAF70 комплекса TFI - ID РНК-полимеразы II, а также с транскрипционным кофактором р300/ СВР. Кроме того, этот домен участву ет в белок-белковых взаимодействиях, регулирующих стабильность молекулы р53. И, наконец, в нем расположено несколько остатков серина и треони на, фосфорилирование которых регу лирует активность р53.
Центральный домен р53 (амино кислоты 120—290) непосредственно узнает и связывает специфические по следовательности Д Н К регулируемых генов, так называемые р53-респонсив- ные элементы, состоящие из располо женных друг за другом последователь ностей с обобщенной структурой типа PuPuC(A/T)(A/T)GPyPyPy (Pu - пу рин, Ру — пиримидин). Именно в этом ДНК-связывающем домене ло кализуется большинство точечных му таций, обнаруживаемых в различных опухолях человека (см. схему 3.8).
Далее идут участки, ответственные
за ядерную локализацию (аминокис лоты 305—323) и димеризацию/тетрамеризацию молекул р53 (аминокисло ты 323—356). С-концевой участок р53 (аминокислоты 363—392) представля ет собой так называемый ингибиторный домен. В немодифицированном состоянии он препятствует посадке ДНК-связывающего домена на специ фическую последовательность регули руемого гена. Фосфорилирование и ацетилирование его определенных сайтов вызывают изменения конформации белковой молекулы и переход тетрамеров р53 из неактивного (ла тентного) состояния в активное. В ре зультате ДНК-связывающие домены освобождаются от блокирующего влияния ингибиторных доменов и приобретают способность садиться на р53-респонсивные элементы. Таким образом, к респонсивным генам при влекаются базовые факторы транс крипции, связывающиеся с N-конце вым участком р53, и стимулируется синтез Р Н К генов-мишеней.
Помимо повышения транскрипции генов, содержащих специфические респонсивные элементы, белок р53 обладает также и рядом других актив ностей. В частности, он способен по давлять транскрипцию многих других генов, например протоонкогенов BCL2, J U N и FOS, гена фибронектина и т. д. В основе такой трансрепрес сии лежит несколько механизмов: связывание и секвестрация активиро ванным р53 ряда базовых факторов транскрипции (р300/СВР, ТВР, CBF); способность связывать и рекрутиро вать к определенным генам гистоновые деацетилазы (HDAC), ремоделирующие хроматин, и т. д. Кроме того, р53 связывается с белками, вовлечен
ными |
в |
репликацию |
или репарацию |
Д Н К , |
и |
как следствие |
модулирует эти |
процессы. Так, взаимодействуя с бел ком RP-A, он ингибирует его способ ность активировать ДНК-полимеразы α и δ, результатом чего является по давление репликации Д Н К . Связывая компоненты комплекса TFIIH (ERCC2, ERCC3 и др.), р53 активиру-
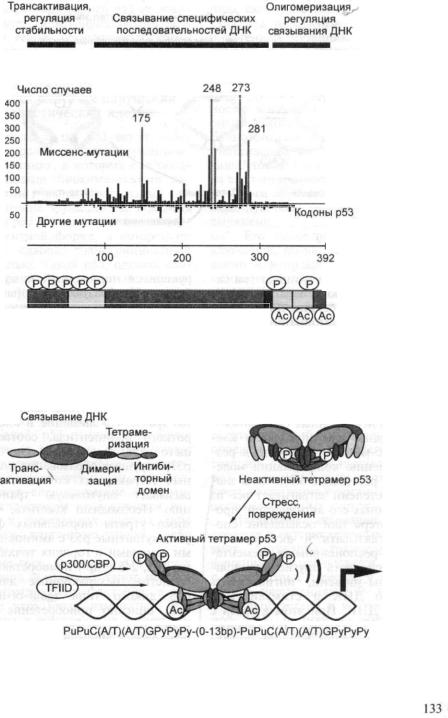
С х е м а 3.8. Функциональные домены р53 предполагаемой моде
ли приобретения белком транскрипционно активной конформации и частота встречаемости в новообразованиях человека мутаций в разных участках молекулы р53
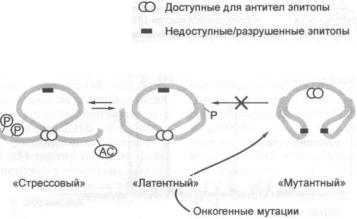
С х е м а 3.9. Различные конформационные состояния р53, распо знаваемые специфическими антителами. Онкогенные мутации вызывают необратимый переход молекулы в денатурированное состояние, при котором открывается ранее недоступный эпитоп и, наоборот, исчезают некоторые ранее доступные эпитопы
ет его функцию и стимулирует тем са мым эксцизионную репарацию Д Н К .
Связывание |
р53 с белком Rad51 ведёт |
||
к стимуляции рекомбинаций |
Д Н К |
и |
|
повышению |
эффективности |
репара |
|
ции двунитевых разрывов Д Н К . |
На |
||
участие р53 |
в репарации Д Н К указы |
||
вает также и его способность прояв лять активность 3'—5'-экзонуклеазы и узнавать участки одноцепочечной Д Н К и/или неспаренные основания.
Характерные для опухолевых кле ток миссенс-мутации приводят к рез кому изменению конформации моле кулы белка р53 (схема 3.9), что в значительной степени затрагивает все из вышеуказанных его активностей: происходит потеря или ослабление способности связывать и активировать гены с р53-респонсивными элемента ми, репрессировать другие специфи ческие гены-мишени, ингибировать репликацию Д Н К и стимулировать репарацию Д Н К . При этом в связи с тем, что р53 образует тетрамерные комплексы, мутации в одном аллеле гена р53 вызывают инактивацию и продукта второго, неповрежденного
аллеля. Дело в том, что коэкспресси-
рующиеся нормальный и мутантный белки р53 образуют неактивные гетеромерные комплексы. Таким образом, мутантный белок ингибирует функ ции нормального белка р53 по доми нантно-негативному механизму. Повидимому, именно эта особенность мутантных р53 в значительной мере ответственна за их онкогенный потен циал. В пользу этого свидетельствует тот факт, что введение в клетки ко роткого полипептида, соответствую щего олигомеризационному домену р53, нарушает образование полноцен ных тетрамерных комплексов р53 и вызывает опухолевую трансформа цию. Необходимо заметить, что, по мимо утраты нормальных функций р53, мутантные р53 с аминокислотны ми заменами в горячих точках (кодоны 175, 248 и др.) приобретают новые свойства, нехарактерные для белка р53 дикого типа (gain-of-function). Так, описано приобретение мутантными р53 способности активировать промоторы протоонкогенов MYC и ERB1 , антиапоптотического гена BGL1 из семейства Вс1-2, гена MDR1, детерминирующего множественную
134
лекарственную устойчивость клеток, и т. д. Предполагается, что это обуслов лено способностью некоторых мутантных р53 связывать белки, в част ности другие факторы транскрипции, с которыми нормальный р53 не взаи модействует, и модифицировать экс прессию генов, регулируемых этими транскрипционными факторами.
3.2.2.3. Физиологические функции р53 и их нарушения в неопластических клетках
Молекулы белка р53 могут нахо диться в различных конформационных состояниях, в которых они обла дают разными биохимическими ак тивностями и выполняют разные фи зиологические функции. В обычных условиях р53 находится в так называе мой латентной форме, в которой он обладает слабой транскрипционной активностью. Такой р53, однако, свя зывает белки репарационной машины (см. выше), проявляет активность 3'— 5'-экзонуклеазы и стимулирует реком бинацию и репарацию Д Н К . При раз личных стрессах и внутриклеточных повреждениях происходят посттранс ляционные модификации, в частности фосфорилирование и ацетилирование определенных аминокислот молекулы р53, определяющие ее переход в так называемую стрессовую конформацию. Такой р53 значительно более стабилен (т. е. резко увеличивается его количество в клетке) и эффектив но транс-активирует и/или транс-ре- прессирует специфические гены-ми шени, следствием чего является ин-
!' дукция в аномальных клетках либо остановки клеточного цикла, либо апоптоза.
Кроме того, активация р53 ведет к изменению экспрессии генов некото рых секретируемых факторов, в ре зультате чего могут изменяться раз множение и миграция не только по врежденной, но и окружающих кле ток. При этом, находясь в стрессовой конформации, р53 в значительной степени утрачивает активность, сти-
мулирующую рекомбинацию и/или репарацию Д Н К . р53 дикого типа в дополнение к латентной и стрессовой может временно приобретать и так называемую мутантную конформацию, сходную с той, в которую моле кула р53 необратимо переходит при онкогенных мутациях. Транзиторный переход р53 в мутантную конформацию происходит при воздействии оп ределенных цитокинов и/или морфогенов ( P D G F , тромбопоэтин, ретиноевая кислота и др.). Биологический смысл такого перехода пока не ясен. Возможно, он заключается в полной инактивации ростингибирующих ак тивностей р53 и/или изменении набо ра его генов-мишеней.
Таким образом, р53 играет важную охранную роль, являясь, по образному выражению D. Lane, "стражем гено ма". Его повседневная функция за ключается, по-видимому, в распозна вании и исправлении ошибок, неиз менно возникающих в ходе реплика ции Д Н К . При массивных поврежде ниях Д Н К , других внутриклеточных нарушениях или угрозе их возникно вения происходит переключение функций р53 (схема 3.10): приобретая транскрипционные активности и из меняя экспрессию генов-мишеней, он вызывает либо остановку размноже ния аномальных клеток (временную, для устранения повреждений, или не обратимую), либо их гибель (факторы, определяющие судьбу клетки при ак тивации р53 будут рассмотрены ни же). В результате устраняется возмож ность накопления в организме генети чески измененных клеток.
Механизмы активации р53 при стрессах и внутриклеточных поврежде ниях. Активация транскрипционных функций р53 наблюдается при самых разнообразных стрессах и внутрикле точных нарушениях: УФ- и гама-облуче- нии, присутствии в клетке разорван ной Д Н К , понижении внутриклеточ ного пула нуклеотидов, ингибировании Д Н К - и РНК-полимераз, гипер экспрессии онкогенов, вирусной ин фекции, гипоксии, оксидативном
135

С х е м а 3.10. Охранные функции р53
А
А — функции "латентной" и "стрессовой" форм р53; Б — факторы, вызывающие транс крипционную активацию р53, гены-мишени активированного р53 и вызываемые изме нениями их экспрессии биологические эффекты
стрессе, гипо- и гипертермии, различ ных нарушениях клеточной архитек туры (увеличении числа ядер, измене ниях цитоскелета и адгезии) и т. д.
Ключевую роль в стабилизации белка р53 и повышении его транс крипционной активности играют изменения взаимодействия р53 с бел ком-ингибитором Mdm2, ген которо го является потенциальным онкоге ном (см. раздел 3.1). Белок Mdm2 свя зывается с N - концом молекулы р53 и,
обладая активностями ЕЗ убиквитинлигазы, стимулирует убиквитинизацию и как результат протеосомную деградацию белка р53, поэтому в нор ме уровень экспрессии р53 очень не велик, а время его жизни составляет всего около 30 мин. Кроме того, свя зываясь с N-концевым участком р53 в районе домена, взаимодействующего с базовыми факторами транскрипции, Mdm2 подавляет способность р53 транс-активировать гены-мишени.
136
При внутриклеточных нарушениях, в частности при повреждениях ДНК, происходит фосфорилирование р53 по сайтам (Serl5, Ser20, Ser33), располо женным в районе связывания с белком Mdm2. Такое фосфорилирование осу ществляют специфические киназы (ATM, ATR и их мишени — чекпойнткиназы С Н К 1 , СНК2), которые акти вируются в ответ на разнообразные на рушения структуры Д Н К (см. раздел 3.2.10.1). Кроме того, ATM фосфорилирует и белок Mdm2. В результате блокируется связывание р53 с Mdm2, что вызывает стабилизацию молекул р53 и повышение их транскрипцион ной активности. При некоторых других внутриклеточных изменениях, напри мер при экспрессии в клетке активиро ванных онкогенов RAS, также наблю дается нарушение взаимодействия р53 и Mdm2, но происходит оно из-за по вышения экспрессии белка pARF, продукта альтернативной рамки счи тывания гена INK4a (см. раздел 3.2.3). Белок pARF обладает способностью связываться либо с N-концевым уча стком р53, либо с белком Mdm2, пре пятствуя, таким образом, их непо средственному взаимодействию. Ин тересно, что ген MDM2 сам является транскрипционной мишенью активи рованного р53. В результате устанав ливаются регуляторная петля, стиму лирующая деградацию белка р53 по сле окончания действия факторов, вызывающих его фосфорилирование или связывание с белком pARF.
Важную роль в приобретении моле кулами р53 конформации, способной транс-активировать гены-мишени, иг рают также модификации С-концевого участка, а именно ацетилирование его
определенных аминокислотных |
остат |
||
ков. При |
повреждениях Д Н К |
и экс |
|
прессии |
активированного |
онкогена |
|
RAS эти события инициируются связы |
|||
ванием освобождающегося |
от |
Mdm2 |
|
N-концевого участка р53 с базальным |
|||
фактором |
транскрипции |
р300/СВР, |
|
ацетилирующим сначала |
ингибитор- |
||
ный домен р53 по лизинам 373 и 382, а затем (после связывания р53 с респон-
сивными элементами) и белки хрома тина в области генов-мишеней. Таким образом, последовательные посттранс ляционные модификации N-концевого
иС-концевого участков р53 вызывают увеличение количества белка р53 в клетке, приобретение им способности связывать р53-респонсивные элементы
ирекрутировать к генам-мишеням ба зовые факторы транскрипции (компо ненты комплекса TFIID РНК-полиме- разы II и гистоновые ацетилазы р300/ СВР, деконденсирующие хроматин), стимулируя тем самым транскрипцию их мРНК .
При некоторых стрессах, в частно сти при гипоксии, наблюдаются по сттрансляционные изменения р53, вы зывающие его переход не к классиче ской стрессовой конформации, а к ее варианту. Такой р53 не транс-активи- рует гены, содержащие р53-респонсив- ные элементы, но подавляет транс крипцию других генов-мишеней. Эта так называемая репрессионная форма также фосфорилирована по N-концу, но ее С-концевой участок не ацетилирован и связывает репрессионные ком плексы Sin3/HDAC, вызывающие кон денсацию хроматина генов-мишеней.
Гены-мишени р53 и их функции. В
настоящее время, помимо Mdm2, обеспечивающего регуляцию самого р53 по принципу обратной связи (см. выше), идентифицировано более 100 генов, являющихся мишенями транс крипционных активностей р53. Они могут быть разделены на несколько групп, исходя из их физиологических функций (см. схему 3.10).
Первую группу составляют гены, продукты которых регулируют клеточ
ный цикл. Важнейшим из них являет ся белок р21Wan/Cipl — ингибитор цик
линзависимых киназ из семейства Cip/Kip (см. раздел 2.2.1). Повышение его экспрессии вызывает остановку клеточного цикла в поздней фазе G 1 , что обусловливается связыванием им комплексов циклин E/cdk2, подавле нием их способности фосфорилировать белки семейства pRb и освобож дать транскрипционные факторы E2F
137
(см. раздел 3.2.1.2). Дублирующим ме ханизмом остановки перехода из G1 в S является подавление активирован ным р53 транскрипции гена DP1 — транскрипционного фактора, который связывается с E2F и образует актив ный комплекс, собственно и активи рующий синтез продуктов, необходи мых для входа в фазу S. Заслуживает также внимания способность р53 по вышать экспрессию гена Siah1, про дукт которого стимулирует деграда цию бета-катенина — транскрипционно го фактора, активирующего транс крипцию гена циклина D и онкогена MYC (см. раздел 3.1.3.4), что вносит дополнительный вклад в индукцию остановки клеточного цикла в G 1 . Следует заметить, что р53 может по давлять активность Cdk2 не только в результате изменения своих транс крипционных функций, но и за счет белок-белковых взаимодействий, а именно непосредственного связыва ния циклина Н — компонента киназного комплекса САК, осуществляю щего активирующее фосфорилирова ние циклинзависимых киназ.
Идентифицирован также ряд ге нов-мишеней р53, продукты которых вызывают остановку в фазе G2 (за держка в ней наблюдается в случае, когда р53 активировался уже после того, как клетка прошла G1-чек- пойнт, или в клетках с инактивированным Gl-чекпойнтом). Активиро ванный р53 подавляет функцию ком плекса циклин B/Cdc2, играющего ключевую роль в переходе из G2 в ми тоз, по нескольким механизмам. Вопервых, он транс-активирует ген 14- З-З-о, белковый продукт которого связывает и секвестрирует комплексы циклин B/Cdc2 в цитоплазме, не да вая возможности им попасть в ядро, где они и должны проявлять свою ак тивность. Во-вторых, он транс-акти- вирует ген GADD45, белковый про дукт которого обладает способностью связывать Cdc2, разрушая таким обра зом комплексы циклин B/Cdc2. В- третьих, р53 репрессирует транскрип цию генов циклина В и Cdc2, что
уменьшает синтез их продуктов. Сле дует заметить, что, как и в случае ос тановки клеточного цикла в G1, за держка в G2 при повреждениях ДНК наблюдается и в клетках с инактивированным р53: она происходит в ре зультате подавления функции фосфатаз Cdc25 (Cdc25A при остановке в G1 и Cdc25C при остановке в G2), акти вирующих соответствующие циклинзависимые киназы, однако в клетках с нарушенной функцией р53 происхо дит лишь кратковременная задержка в чекпойнтах, а активация р53 обеспечи вает длительную остановку клеточно го цикла, предотвращающую размно жение вплоть до исправления дефекта.
Следующая группа р53-регулируе- мых генов кодирует белки, индуци рующие апоптоз. При этом р53 кон тролирует синтез компонентов обоих основных путей индукции апоптоза: и митохондриального, и стимулируемо го "рецепторами смерти" (см. раздел 2.2.4). Так, он регулирует активность белков семейства Bcl-2, репрессируя ген антиапоптотического белка Bcl-2 и активируя гены проапоптотических белков Вах, Puma и Noxa. Повышение проницаемости митохондриальной мембраны для цитохрома С и белка AIF достигается и транс-активацией гена р53А1Р1 (его продукт локализует ся в митохондриальной мембране и уменьшает мембранный потенциал) и гена PIG3 (кодирует оксиредуктазу, которая вовлечена в образование ра дикалов кислорода, повреждающих мембраны митохондрий и стимули рующих выброс их содержимого). Стимуляция апоптоза, запускаемого рецепторами смерти, достигается транс-активацией генов двух из таких рецепторов — Fas и Killer/DR5 (ре цептор для TRAIL). Еще одной такой мишенью является ген недавно иден тифицированного белка Pidd, кото рый содержит домен смерти и при ги перэкспрессии индуцирует апоптоз. Выявлены и другие р53-респонсивные
гены |
(IGF-BP3, |
PAG608, |
р85, циклин |
G), |
продукты |
которых |
стимулирует |
апоптоз, однако |
механизмы их апоп- |
||
138
тогенного действия пока недостаточно исследованы. Кроме того, р53 может индуцировать апоптоз и через другие механизмы, не связанные с его спо собностью изменять экспрессию ге нов-мишеней. Так, мутантные р53, ут ратившие транскрипционную актив ность (в результате делеции С-конце вого участка или мутаций в кодонах 22—23), сохраняют тем не менее спо собность индуцировать апоптоз в не которых (но не во всех) типах клеток. Предполагается, что этот эффект обу словлен белок-белковыми взаимодей ствиями р53. Таким образом, при по вышении функциональной активно сти р53 может происходить активация сразу многих путей индукции апопто за, что, по-видимому, обеспечивает надежность его реализации.
В связи с тем что р53 контролирует активность генов, продукты которых способны вызвать как остановку кле точного цикла в различных его фазах, так и апоптоз, возникает вопрос: от чего зависит выбор судьбы клетки при активации р53? Выяснилось, что он определяется множеством факторов: гистогенетическим типом клеток (на пример, в нормальных фибробластах, как правило, наблюдается остановка клеточного цикла, тогда как в лимфо цитах — апоптоз), степенью актива ции р53 (с увеличением уровня его экспрессии повышается вероятность апоптоза), функциональной активно стью сигнального пути pRb-E2F (в фибробластах с инактивированным pRb или гиперэкспрессированным E2F наблюдается не остановка в G l , а апоптоз) и т. д. Недавно обнаружено, что еще одним фактором, определяю щим выбор между остановкой клеточ ного цикла и апоптозом, является ха рактер модификации самих молекул р53 и/или их белок-белковых взаимо действий. Так, р53, фосфорилированный по Serl5/20 и ацетилированный
по С-концу, способен транс-актви- ровать ген p2lWaf/Cip1 и вызывать оста
новку в G 1 , тогда как дополнительное фосфорилирование по Ser46 придает ему способность транс-активировать
наряду с геном р21Waf/Cip1 и ген белка р53А1Р1, а в этом случае уже наблю дается апоптоз, причем вероятность фосфорилирования Ser46 повышается с увеличением интенсивности повре ждений ДНК . Кроме того, способ ность р53 избирательно транс-активи- ровать проапоптотические гены (ВАХ и др.) увеличивается при его связыва нии с белками семейства ASPP (ASPP1 и ASPP2), потеря экспрессии которых характерна для значительной части случаев рака молочной железы.
Третьей большой группой геновмишеней р53 являются гены, продук ты которых регулируют морфологию и/или миграцию клеток (см. схему 3.10). Так, р53 транс-активирует гены
обоих представителей |
семейства |
рас |
|
сеивающих |
(Scatter) |
факторов |
— |
H G F / S F и |
HGF1/MSP, а также |
ген |
|
одного из членов семейства эпидермальных факторов роста H B - E G F (гепаринсвязывающий EGF); продукты всех этих генов являются одновремен но и митогенами, и мотогенами. При этом р53 транс-активирует также ге
ны рецепторов |
этих факторов — |
H G F / S F - R (Met) |
и EGF-R. р53-Рес- |
понсивными являются также гены хемокина фракталкина, гладкомышечного альфа-актина, коллагенов III и VII типов, ингибитора плазминогена PAI- 1. В то же время р53 репрессирует ге ны фибронектина и металлопротеиназы I типа. Физиологическое значение такой регуляции пока не установлено. Возможно, оно заключается, по край ней мере частично, в привлечении к клеткам с активированным р53 окру жающих клеток определенных типов для ремоделирования/восстановления структуры ткани в месте возмож ного апоптоза. Не исключено участие такой регуляции и в процессах мор фогенеза.
В особую группу генов-мишеней р53 можно выделить гены, контроли рующие ангиогенез. Ключевую роль в неоангиогенезе играет V E G F (Vascular Endothelial Growth Factor; стимулиру ет размножение и миграцию эндотелиоцитов), экспрессия которого по-
139
вышается при гипоксии или актива ции некоторых онкогенов (см. главу 9). р53 репрессирует транскрипцию как гена VEGF, так и гена HIF-1 (Hy poxia Inducing Factor 1) — транскрип ционного фактора, обеспечивающего повышение экспрессии V E G F и его рецепторов в ответ на уменьшение со держания кислорода (кроме этого, HIF-1 изменяет экспрессию генов, контролирующих транспорт глюкозы и гликолиз, что обеспечивает адапта цию клеток к условиям гипоксии). Одновременно р53 может транс-акти- вировать гены белков, ингибирующих ангиогенез, — тромбоспондинов (Tsp)-l, -2 (связывая специфические рецепторы на поверхности эндотелиоцитов, они вызывают в них апоптоз) и BAI-1. Таким образом, клетки с акти вированным р53 хуже переносят не достаток кислорода, перестают секретировать VEGF и начинают секретировать ингибиторы ангиогенеза, что препятствует образованию новых со судов. Данные функции являются, повидимому, еще одной составляющей опухольсупрессирующего действия р53, так как они предотвращают адап тацию к гипоксии и прорастание со судов в центр опухоли.
Выявлено еще несколько десятков генов-мишеней р53. Среди них следу ет отметить ген каталитической субъ единицы теломеразы (TERT), кото рый репрессируется р53 (таким обра зом, р53 участвует по-видимому, и в обеспечении репликативного старе ния клеток — см. раздел 2.2.3 и 3.5). По-видимому, р53 принимает участие и в процессах созревания клеток, так как некоторые из транс-активируе- мых им генов кодируют белки репер туара той или иной дифференцировки (мышечная креатинкиназа и др.).
Последствия нарушений функции р53. Характерные для опухолевых кле ток аномалии р53 отменяют или ос лабляют все важнейшие функции опу холевого супрессора р53. Делеция обоих аллелей гена р53, т. е. его пол ная инактивация, вызывает ослабле ние G 1 - и С2-чекпойнтов клеточного
цикла, подавление индукции апопто за, уменьшение эффективности репа рации Д Н К , более эффективную адаптацию к гипоксии и стимуляцию неоангиогенеза, ослабление контроля за длиной теломер, ингибирование дифференцировки и другие характер ные свойства неопластической клетки (см. главу 2). Особо следует отметить возникновение в клетках с инактивированным р53 сильной генетической нестабильности, являющейся мотором дальнейшей опухолевой прогрессии. Потеря функциональной активности р53 значительно увеличивает темп по явления размножающихся клеток с самыми разными генетическими ано малиями — измененным числом и пе рестройками хромосом, генными му тациями, амплификацией отдельных участков генома.
Сходные последствия наблюдаются
ипри самых частых в новообразова ниях человека аномалиях р53 — мис- сенс-мутациях, ведущих к синтезу не активного белка, обладающего доми нантно-негативным эффектом в отно шении продукта неповрежденного аллеля. Следует заметить, однако, что степень проявления доминантно-не гативного эффекта мутантного р53 варьирует в зависимости от конкрет ной аминокислотной замены, типа клеток и ингибируемой функции р53 дикого типа. В связи с этим нередко селективное преимущество получают клоны клеток, в которых в результате дополнительных генетических собы тий происходит делеция или мутация
ивторого аллеля гена р53. В то же время опухолевые клетки, как прави ло, сохраняют экспрессию хотя бы од ного мутантного аллеля гена р53. Оче видно, это можно объяснить тем, что появляющиеся в результате миссенсмутаций новые активности мутантных р53 вносят дополнительный вклад в повышение онкогенного потенциала клетки. Так, приобретая способность транс-активировать онкоген МYС, мутантный р53 вызывает, очевидно, более сильные нарушения регуляции клеточного цикла, чем те, которые на-
140
блюдаются при делениях гена р53. Новые активности мутантных белков р53 ответственны также за дополни тельное ослабление индукции апопто за и развитие устойчивости к дейст вию химиопрепаратов. В основе по давления апоптоза лежит несколько механизмов: т/?анс-активация мутант ный р53 гена антиапоптотического белка Bagl (член семейства bcl-2), способность мутантных р53 связывать и инактивировать гомолог р53, белок р73 (см. следующий раздел) и т. д. Возникновение устойчивости к опре деленным противоопухолевым цитостатикам может быть связано также со способностью некоторых мутантных р53 повышать транскрипцию гена MDR1 (Multi-Drug Resistance 1 — см. раздел 13.1.1) и гена дУТФазы, про дукт которого блокирует действие 5- фторурацила и ряда других антимета болитов. Следует заметить, что харак терные для опухолей человека мута ции дифференциально влияют на воз никновение вышеуказанных активно стей. Так, замены в кодонах 175 и 248 придают способность активировать ген дУТФазы, тогда как мутации в ко доне 273 не вызывают приобретения такого свойства, поэтому использова ние мутаций р53 в качестве критерия для предсказания чувствительности к той или иной химиотерапии может быть основано лишь на точной иден тификации аминокислотных замен, а не на гистохимической детекции "нормального" или "мутантного" р53 с помощью конформационно-специфи- ческих антител.
Таким образом, мутации и другие изменения активности р53 вызывают одновременное появление целого на бора характерных свойств неопласти ческой клетки (см. главу 2), таких как понижение чувствительности к раз личным ростсупрессирующим сигна лам (в том числе генерируемым пер манентной экспрессией активирован ных онкогенов), иммортализация, по вышение способности выживать в не благоприятных условиях, генетиче ская нестабильность, стимуляция нео-
ангиогенеза, блокирование клеточной дифференцировки и т. д. Это, очевид но, и является объяснением такой частой встречаемости мутаций р53 в самых разных новообразованиях — они позволяют за один шаг преодо леть сразу несколько этапов опухоле вой прогрессии, причем мутации р53 могут являться как инициальным со бытием (синдром Ли—Фраумени) или детерминировать начальные этапы канцерогенеза, так и возникать и от бираться уже в ходе роста опухоли, обеспечивая приобретение новых аг рессивных свойств и устойчивости к терапии.
3.2.2.4. Гомологи р53: р6З и р73
Недавно были обнаружены два ге на — р6З и р73, продукты которых имеют достаточно высокую степень гомологии с белком р53 в участках транс-активационного, ДНК-связы- вающего и олигомеризационного до менов, но которые значительно отли чаются от него по С-концу. В отличие от гена р53, кодирующего по существу один белковый продукт, подвергаю щийся, правда, самым разным по сттрансляционным модификациям, гены р6З и р73 продуцируют по не сколько белков. Дело в том, что м Р Н К каждого из них может синтези роваться с двух промоторов и далее подвергаться альтернативному сплай сингу. В результате образуется по 6 изоформ белков р6З и р73, обладаю щих (ТА-формы) или не обладающих (дельтаN-формы) транс-активационным доменом. При этом формы, содержа щие транс-активационный домен, способны активировать р53-респон- сивные гены и вызывать, в частности, апоптоз. Кроме того, ТА-формы р6З и р73 могут транс-активировать и ряд других генов, содержащих близкие по структуре респонсивные элементы, которые не являются тем не менее мишенями р53. В частности, они яуюис-активируют гены белков Jagl и Jag2 — лигандов для рецепторов
141
Notch, активация которых играет ключевую роль в судьбе клетки при выборе направления ее дифференцировки.
Существует еще ряд отличий меж ду р53 и его гомологами. Так, если р53 экспрессируется в клетках практиче ски всех тканей, то его гомологи экспрессируются только в некоторых ти пах клеток. Например, р63 преимуще ственно экспрессируется в эмбрио нальных клетках, а также стволовых и недифференцированных эпителиаль ных клетках взрослого организма, причем в виде транскрипционно-не- активных ΔΝ-форм. Предполагается, что экспрессия ΔΝ-форм р63 обеспе чивает недифференцированное со стояние клетки. При нокауте гена р63 у мышей наблюдается пренатальная или постнатальная гибель эмбрионов вследствие полного отсутствия кожи и других эпителиальных тканей (пред полагается, что это связано с прежде временной дифференцировкой ство ловых клеток эпителия и исчерпанием их запаса к моменту рождения живот ных). Далее, если р53 активируется в ответ на самые разные стрессы, то его гомологи — только на некоторые из них, причем за счет совершенно дру гих механизмов. Наконец, если р53 выполняет функции опухолевого супрессора и для новообразований ха рактерна его инактивация, то его го мологи не имеют таких функций. Об этом свидетельствуют две группы фактов. Во-первых, у мышей гомози готный нокаут гена р73 не вызывает повышения частоты возникновения опухолей (нокаут гена р63 несовмес тим с жизнью эмбрионов — см. вы ше). Во-вторых, в опухолях человека не выявляется потеря экспрессии или мутации генов р63 и p73. Наоборот, в них нередко отмечается повышение экспрессии этих белков, причем, как правило, транскрипционно-неактив- ных ΔΝ-форм (т. е. р63 и р73 являют ся скорее протоонкогенами). В связи с этим приобретает популярность ги потеза, согласно которой ΔΝ-формы белков р63 и р73 функционируют как
142
природные ингибиторы р53, подав ляющие его функцию по доминантнонегативному механизму. Действитель но, их транскрипционно-неактивные тетрамеры могут конкурировать с р53 за места посадки на Д Н К генов-ми шеней, а мономеры/димеры — секве стрировать р53 путем связывания его молекул и образования неактивных комплексов. Это не исключает, одна ко, что при нарушениях функции р53 происходит активация транскрипци онной функции его гомологов (пока зано, что р53 репрессирует транс-гк- тивационную функцию белка р73), которая может частично компенсиро вать утрату функции р53 и обеспечи вать, например, нормальное развитие мышей с гомозиготным нокаутом гена ρ53. Эти предположения, как и другие аспекты биологических функций го мологов р53, требуют дальнейшего ис
следования
3.2.3 Продукты гена INK4a - p16INK4a и pARF регулирующие активность pRb и p53
Следующим после р53 по частоте изменений в различных новообразо ваниях человека является ген ΙΝΚ4а, расположенный в коротком плече 9-й хромосомы (сегмент 9р21). Его ис ключительной особенностью являет ся одновременное кодирование двух
негомологичных ядерных белков — pl61NК4a и pARF (продукты альтерна
тивных рамок считывания), каждый из которых выполняет супрессорные функции (схема 3.11).
pl61NK4a связывает циклинзависимые киназы cdk4 и cdk6 (см. раздел 2.2.1) и препятствует образованию их функционально-активных комплексов с циклинами D, которые, фосфорилируя pRb, инициируют вход в S-фазу клеточного цикла (см. раздел 3.2.1.2). pARF обладает способностью стаби лизировать и активировать белок р53, нарушая его взаимодействие с белком Mdm2 (см. раздел 3.2.2.3). Кроме того, у него выявлены и р53-независимые
-

С х е м а 3.11. Ген INK4a кодирует два негомологичных белка,
выполняющих разные функции
функции: он может также непосредст венно взаимодействовать с основной мишенью pRb — белком E2F и блоки ровать его активность. Кроме того, он понижает стабильность транскрипци онного фактора HIF-1 (см. разделы 3.2.2.3 и 3.2.7), связывая его а-субъе-
диницу.
Активность pl6INK4a и pARF повы шается при экспрессии ряда вирусных
(Е1А) или клеточных (RAS, RAF, МУС
и др.) онкогенов. Такой эффект обу словлен присутствием в гене INK4a респонсивных элементов для транс крипционного фактора E2F, активи руемого многими онкогенами. Таким образом, нормальное функционирова ние продуктов гена INK4a эффектив но предотвращает дальнейшее раз множение клеток, в которых про изошла активация какого-либо из представителей большой группы он когенов. Кроме того, экспрессия pl6INK4a повышается при образовании межклеточных контактов, что обеспе чивает контактное торможение раз множения клеток (см. раздел 2.1).
У трансгенных мышей с гомози готной ??? нокаутом гена INK4a, вы зывающим потерю экспрессии обоих его белковых продуктов, наблюдается высокая частота возникновения в мо-
лодом возрасте различных новообра зований — преимущественно фибросарком и лимфом (так же как и у мы шей с инактивированным р53). Инте ресно, что практически такая же кар тина наблюдается и у мышей с пере стройкой гена INK4a, приводящей к потере экспрессии только белка
pARF. В отличие от этого у мышей, утративших экспрессию pl6INMa, но
сохранивших экспрессию pARF, нет существенного повышения частоты возникновения опухолей (отмечается лишь редкое возникновение меланом, не наблюдаемое в родительских лини ях мышей). В культурах in vitro мыши ные фибробласты, не экспрессирующие оба продукта гена INK4a, как и фибробласты, в которых инактивирован только pARF, легко трансформи руются онкогенами семейства RAS. В то же время для RAS-индуцированной трансформации клеток, не экспрессирующих только pl6INK4a, необходимо дополнительное действие коопери рующих онкогенов, таких как МYС или Е1А (см. раздел 3.1), подобно то му, как это наблюдается в случае нор мальных клеток.
У людей терминальные мутации в одном из двух аллелей гена lNK4a ассоциированы с наследственной пред-
143
расположенностью к развитию меланом кожи (синдром диспластических невусов). Часть из этих мутаций инактивирует только pl61NK4a, не нарушая функцию pARF. В связи с этим пред полагается, что синдром диспластиче ских невусов связан с нарушением функции именно pl61NK4a. В клетках наследственных и спорадических меланом обнаруживают изменения обо их аллелей гена INK4a, т. е. данный ген ведет себя как классический опу холевый супрессор. Чаще всего вторая (соматическая) мутация представляет
собой делецию гена INK4a, ведущую к инактивации как pl6ІNK4a, так и pARF.
Мутации, делеции и метилирование гена INK4a, вызывающие потерю экс прессии одного или обоих его белко вых продуктов, часто наблюдаются не только в наследственных и спорадиче ских меланомах, но и в большой груп пе других ненаследственных новооб разований: раке поджелудочной желе зы, пищевода, желчных путей, моче вого пузыря, Т- и В-клеточных острых лимфолейкозах, мезотелиомах, анапластических астроцитомах, глиобластомах и др.
3.2.4. Опухолевый супрессор PTEN регулирует
клеточный цикл и апоптоз, модулируя сигнальный путь PI3K-PKB/Akt
К опухолевым супрессорам, часто поражаемым в различных новообразо ваниях человека, относится и ген PTEN. Его терминальные мутации вы зывают болезнь Коудена, заключаю щуюся в наследственном предраспо ложении к развитию гамартом, чаще всего в мозге, молочной и щитовид ной железах. Инактивация PTEN характеризует также и значительную часть различных ненаследственных опухолей — глиомы, менингиомы, меланомы, раки почки, матки, молочной и предстательной желез. При этом на начальных стадиях заболевания выяв ляется, как правило, делеция только одного из аллелей гена PTEN, тогда
как в опухолях, находящихся на позд них стадиях развития, чаще инактивированы оба аллеля. У трансгенных мышей с нокаутом одного аллеля гена PTEN отмечается развитие в молодом возрасте различных новообразований — аденокарцином кишечника, лейкозов, лимфосарком, терминальных опухо лей и др.
Белковый продукт гена PTEN со стоит из 403 аминокислот и имеет вы сокую степень гомологии с фосфатазами двойной специфичности (дефосфорилируют и тирозиновые, и серии/ треониновые аминокислотные остат ки). Его особенностью является спо собность дефосфорилировать не толь ко белки, но и липиды, в частности фосфатидилинозит (3, 4, 5) трифосфат (PIP-3) — важнейшую мишень фос- фатидилинозитид-З'-киназы (PI3K), участвующую в регуляции клеточного цикла и апоптоза (см. раздел 3.1). Именно эта активность белка PTEN ответственна, по-видимому, за его супрессорную функцию. Об этом свиде тельствует, в частности, тот факт, что большинство мутаций, обнаруживае мых в опухолевых клетках, картиру ются в фосфатазном домене и отменяют дефосфорилирование PIP-3, ингибируя, таким образом, сигнальный путь PI3K-PKB/Akt. В связи с тем что онкоген PKB/Akt подавляет митохондриальный путь индукции апоптоза по нескольким механизмам (ингибирует Bad, активирует bcl-2 и т. д.), по теря функции PTEN делает клетки менее чувствительными ко многим апоптогенным стимулам. В клетках с инактивированным PTEN наблюдает ся также и стимуляция клеточной пролиферации (повышение экспрес сии PKB/Akt увеличивает уровень циклина D). Восстановление функции PTEN в опухолевых клетках приводит в зависимости от клеточного контек ста либо к апоптозу (рак предстатель ной железы), либо к остановке кле точного цикла в G1 (глиобластома, рак почки). Следует заметить, что клетки глиобластомы и рака почки становятся при этом сенсибилизиро-
144
ванными |
к действию дополнительных |
мейства |
трансмембранных |
гликопро- |
||||||||||||||||
апоптогенных |
стимулов. |
|
|
теинов, |
осуществляющий |
адгезион |
||||||||||||||
PTEN участвует, по-видимому, и в |
ные межклеточные контакты типа "зо |
|||||||||||||||||||
регуляции адгезии и миграции клеток. |
на слипания" (zona adhaerence). Его |
|||||||||||||||||||
Его N-концевой домен гомологичен |
внутриклеточный домен связывается с |
|||||||||||||||||||
тензину |
— белку, взаимодействующе |
рядом белков, в первую очередь с р- |
||||||||||||||||||
му в фокальных контактах с актино- |
катенином. В этом случае происходит |
|||||||||||||||||||
вым |
цитоскелетом. |
Дефосфорилируя |
переключение |
функции |
р-катенина: |
|||||||||||||||
тирозиновые |
остатки |
киназы фокаль |
он перестает функционировать в каче |
|||||||||||||||||
ных контактов FAK (см. раздел 3.1 и |
стве |
транскрипционного |
фактора, а |
|||||||||||||||||
главу 7), PTEN может ингибировать |
взаимодействует с актиновыми мик- |
|||||||||||||||||||
образование |
фокальных |
контактов и |
рофиламентами и участвует в регуля |
|||||||||||||||||
распластывание клеток, а также их |
ции |
реорганизации |
цитоскелета. |
Ха |
||||||||||||||||
движение. |
|
|
|
|
|
рактерные для опухолей желудочно- |
||||||||||||||
|
|
|
|
|
|
|
|
кишечного тракта, рака молочной же |
||||||||||||
3.2.5. Е-кадхерин, АРС |
|
лезы, |
яичника и |
ряда других |
новооб |
|||||||||||||||
|
разований изменения |
гена |
Е-кадхери |
|||||||||||||||||
и |
аисин — супрессоры, |
|
||||||||||||||||||
|
на |
(мутации, |
делеции, |
метилирова |
||||||||||||||||
контролирующие |
|
|
||||||||||||||||||
|
|
ние) |
вызывают |
потерю |
экспрессии |
|||||||||||||||
сигнальный путь бета-катенин/ |
||||||||||||||||||||
Cdk/pRb |
|
|
|
|
|
белка или изменение его локализации |
||||||||||||||
|
|
|
|
|
в клетке, что имеет два важных для |
|||||||||||||||
|
|
|
|
|
|
|
|
|||||||||||||
Наследственные |
и |
спорадические |
онкогенеза последствия. Во-первых, |
|||||||||||||||||
опухоли |
желудочно-кишечного |
трак |
нарушаются |
межклеточные |
контакты |
|||||||||||||||
та довольно часто ассоциированы с |
и |
морфогенетические |
реакции клетки |
|||||||||||||||||
мутациями в генах Е-кадхерина, р-ка- |
и, |
во-вторых, |
происходят |
накопление |
||||||||||||||||
тенина, АРС и аксина (см. табл. 3.5). |
р-катенина в ядре и повышение его |
|||||||||||||||||||
Так, |
гемизиготные терминальные му |
транскрипционной |
активности |
(схе |
||||||||||||||||
тации |
|
Е-кадхерина |
|
обусловливают |
ма 3.12). В результате стимулируется |
|||||||||||||||
развитие |
наследственных |
форм |
рака |
размножение |
|
клеток |
|
и |
увеличивается |
|||||||||||
желудка. Врожденные |
мутации одного |
их способность к инвазии. Восстанов |
||||||||||||||||||
из аллелей гена р-катенина или гена |
ление экспрессии Е-кадхерина в рако |
|||||||||||||||||||
АРС ведут к развитию семейного аде- |
вых клетках с помощью введения ген |
|||||||||||||||||||
номатозного |
полипоза |
кишечника (у |
но-инженерных конструкций вызыва |
|||||||||||||||||
большинства |
пациентов |
наблюдаются |
ет резкое замедление пролиферации и |
|||||||||||||||||
мутации АРС, реже встречаются мута |
переход от инвазивного к неинвазив- |
|||||||||||||||||||
ции р-катенина). В основе онкогенно- |
ному фенотипу, причем ростсупресси- |
|||||||||||||||||||
го эффекта всех этих мутаций лежит |
рующий эффект Е-кадхерина обу |
|||||||||||||||||||
повышение транскрипционной функ |
словливается его способностью связы |
|||||||||||||||||||
ции протоонкобелка р-катенина, вы |
вать и секвестрировать р-катенин и не |
|||||||||||||||||||
зывающего активацию |
циклинзависи |
зависит от того, происходит ли при |
||||||||||||||||||
мых киназ и инактивирующего тем |
этом |
восстановление |
|
межклеточных |
||||||||||||||||
самым супрессорную |
функцию |
pRb |
контактов. |
|
|
|
|
|
|
|
|
|
||||||||
(см. раздел 3.2.1). Такая функция р- |
|
В не связанном с Е-кадхерином |
||||||||||||||||||
катенина |
обусловлена |
его способно |
состоянии р-катенин очень нестаби |
|||||||||||||||||
стью транслоцироваться в ядро, свя |
лен. Ключевую роль в регуляции его |
|||||||||||||||||||
зываться |
с транскрипционными |
фак |
времени |
жизни и |
транскрипционной |
|||||||||||||||
торами семейства TCF/LEF и активи |
активности |
играют |
|
опухолевые |
су |
|||||||||||||||
ровать гены, имеющие в своем соста |
прессоры АРС и аксин (см. схему |
|||||||||||||||||||
ве TCF/LEF-респонсивные элементы, |
3.12). АРС связывает одновременно и |
|||||||||||||||||||
в частности ген циклина D1 (см. раз |
р-катенин, и аксин, в результате чего |
|||||||||||||||||||
дел 2.2.1) и онкоген МУС (см. раздел |
образуется сложный комплекс, к ко |
|||||||||||||||||||
3.1). |
|
|
|
|
|
|
|
торому рекрутируется GSK-Зр (кина- |
||||||||||||
Е-кадхерин — представитель се- |
|
за-Зр |
гликогенсинтетазы). |
Фосфори- |
||||||||||||||||
10-7908 Д. Г. Заридзе |
145 |
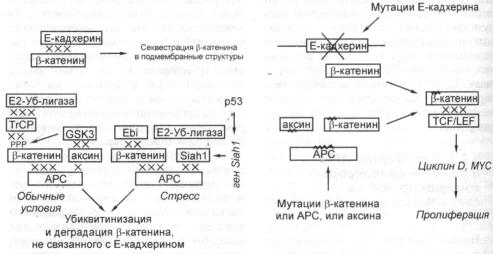
С х е м а 3.12. Регуляция транскрипционной активности р-катенина и ее нарушения в
опухолевых клетках (объяснение в тексте)
Нормальные клетки |
Опухолевые клетки |
лируя определенные сериновые остат ки р-катенина, GSK-Збета индуцирует связывание его с Е2-убиквитин-лига- зой. Убиквитинированный р-катенин направляется в протеосомы, где про исходит его деградация. При стрессах, вызывающих активацию р53, включа ется дополнительный механизм дегра дации бета-катенина (см. схему 3.12). В этом случае происходит повышение экспрессии белка Siahl, который рек рутирует другой тип комплексов Е2- убиквитин-лигазы также и к нефосфорилированному по N-концу бета-кате- нину. Таким образом достигается, повидимому, более эффективная дегра дация не связанного с кадхеринами бетакатенина.
Вероятно, при онкогенных мутаци ях Е-кадхерина свободного бета-катени на становится так много, что системы его деградации не справляются, и часть бета-катенина оказывается в ядре, где он проявляет свою транскрипци онную активность (см. схему 3.12). В клетках новообразований часто реали зуется и другой путь повышения транскрипционных активностей р-ка тенина, связанный с нарушениями ра146
боты систем его деградации. Так, в клетках опухолей толстой кишки, пе чени, предстательной железы часто обнаруживаются либо мутации бета-ка тенина в участках, которые фосфорилируются GSK-3бета и взаимодействуют с убиквитин-лигазой, либо мутации опухолевого супрессора АРС, нару шающие его взаимодействие с бета-кате- нином, либо (значительно реже) мута ции аксина, отменяющие его связыва ние с АРС или GSK-3бета (при этом му тации АРС блокируют работу обеих систем деградации бета-катенина, тогда как указанные мутации самого бета-кате нина или аксина нарушают только од ну из этих систем). В результате всех указанных событий бета-катенин активи рует транскрипцию гена циклина D и протоонкогена МYС, что вызывает стимуляцию клеточной пролифера ции.
Е-кадхерин и АРС являются клас сическими опухолевыми супрессорами: у пациентов с семейными форма ми соответственно рака желудка и аденоматозного полипоза кишечника в опухолевых клетках наблюдается инактивация обоих аллелей данных
генов. Произошедшие еще в половой клетке наследуемые изменения одного из аллелей представляют собой точеч ные инактивирующие мутации, или микроделеции. Второй аллель инактивируется уже в соматической клетке. Подавление активности второго алле ля гена Е-кадхерина нередко связано с его метилированием (см. раздел 3.6), а второй аллель гена АРС чаще всего инактивируется в результате делеции участка длинного плеча 5-й хромосо мы, содержащего данный ген, или ут раты всей 5-й хромосомы. Ключевая роль терминальных мутаций АРС в генезе аденоматозного полипоза тол стой кишки подтверждается тем, что у трансгенных мышей с гомозиготным нокаутом этого гена также развивают ся множественные полипы, причем не только в толстой, но и в тонкой кишке.
Недавно выявлена еще одна важ ная биологическая функция опухоле вого супрессора АРС. Во время мито за он связывается с белками кинетохора и участвует в организации вере тена деления. При инактивации АРС нарушается взаимодействие микро трубочек с кинетохором, результатом чего являются частые ошибки сегрега ции хромосом, т. е. нестабильность генома. Это, вероятно, может служить объяснением более частой встречае мости мутаций АРС по сравнению с мутациями бета-катенина: помимо более эффективного блокирования работы систем деградации бета-катенина, они ведут также и к генетической неста бильности, что придает клеткам до полнительные селективные преиму щества.
3.2.6. Компоненты сигнальных путей TGFбета-Smad
Как опухолевые супрессоры клас сифицированы и некоторые компо ненты сигнальных путей, регулируе мых TGF-бета. Этот цитокин в зависи мости от типа клеток-мишеней и их микроокружения может вызывать ос-
тановку размножения, стимуляцию дифференцировки, а в некоторых слу чаях и апоптоз. Свои антипролиферативные и дифференцировочные эф фекты (они наблюдаются в нормаль ных эпителиальных, эндотелиальных и гемопоэтических клетках) TGF - p реализует по следующему механизму. Его связывание с RII-субъединицей рецептора, обладающей серин-трео- нинкиназной активностью, вызывает рекрутирование и фосфорилирование второй субъединицы рецептора — RI, также являющейся серин-треонино- вой киназой. Основной мишенью ки назы Tбета-RI являются в зависимости от типа клеток белки Smad2 или Smad3 — так называемые рецептор ные Smad. Их фосфорилированные формы образуют комплекс с белком Smad4, который транспортируется в ядро и, формируя еще более сложные комплексы с другими транскрипцион ными кофакторами, функционирует в качестве активаторов транскрипции одних генов и репрессоров других ге нов. В частности, он репрессирует ген МYС и mpанс-активирует гены инги-
биторов циклинзависимых киназ (CKIs) pl5Ink4b, p27Kipl и p21Wafl/Cipl (см.
раздел 2.2.1), что ведет к ингибированию функции cdk4, cdk2 и остановке клеточного цикла в G 1 . При этом ре прессия МYС играет ключевую роль в проведении антипролиферативного сигнала TGF-бета, так как именно она "разрешает" стимуляцию транскрип ции генов CKI комплексом Smad2(3)/ Smad4/Spl (предполагается, что МУС
закрывает места посадки этого ком плекса в генах pl5Ink4b, p27Kipl и p21Wafl/
Cip1). При гиперэкспрессии онкогена МYС, которая характерна для многих новообразований человека, TGF-бета не способен вызвать понижение его экс прессии, достаточное для "разреше ния" стимуляции транскрипции генов CKI, и в результате TGF-бета не оказы вает антипролиферативного действия.
Инактивирующие мутации компо нентов этого сигнального пути, а именно рецептора TGF-бета (TбетаR-II), Smad2 и Smad4, характерны для опу-
147
холей толстой кишки, рака поджелу дочной железы, желчного пузыря, лег кого, а также некоторых других ново образований. Терминальные мутации в одном из аллелей генов TбетаR-II или Smad4 ассоциированы с развитием се мейных форм рака толстой кишки и желудка. Интересно, что у трансген ных мышей с повреждением одного из аллелей гена TpR-II, Smad2 или Smad4 частота развития опухолей не повышается, однако гетерозиготная инактивация одновременно и гена Smad4, и гена АРС резко увеличивает вероятность развития инвазирующих опухолей кишечника. Сходная карти на наблюдается у мышей с нокаутом одного из аллелей гена Smad3. В этом случае наблюдается развитие в моло дом возрасте множественных метастазирующих колоректальных карцином.
3.2.7. Опухолевый супрессор VHL регулирует реакцию на гипоксию и ингибирует ангиогенез
Наследование инактивированного аллеля гена VHL вызывает предраспо ложение к развитию синдрома фон Хиппеля—Линдау. Эта болезнь харак теризуется возникновением опухолей различной локализации — гемангиобластом центральной нервной систе мы, ангиом сетчатки, феохромоцитом, карцином почки, рака поджелудочной железы и др. Инактивация гена VHL характерна и для ряда форм ненаслед ственных опухолей. Так, мутации и/ или гиперметилирование обоих алле лей гена VHL наблюдаются примерно в 80 % светлоклеточных раков почки.
Белковый продукт гена VHL состо ит из 213 аминокислотных остатков и образует комплекс с рядом белков (включая элонгины А, В, С, куллин,
Rbx-1), обладающий |
активностями |
ЕЗ-убиквитин-лигазы. |
Специфиче |
ской мишенью этого комплекса явля ется транскрипционный фактор H I F - 1, гены-мишени которого обеспечива ют адаптацию к гипоксии и стимуля цию ангиогенеза. В присутствии ки
слорода VHL связывает гидроксилированные пролины а-субъединицы HIF - 1, что вызывает убиквитинизацию и деградацию HIF - 1 . При гипок сии HIF-1 не гидроксилируется и не связывается с VHL. В результате со держание HIF-1 в клетке увеличивает ся, что ведет к повышению транс крипции его генов-мишеней, коди рующих VEGF, эритропоэтин, транс портеры глюкозы, гликолитические энзимы, TGF-a и др. В клетках с инактивированным VHL наблюдается перманентное повышение экспрес сии этих генов, что вызывает пониже ние их чувствительности к гипоксии и секрецию ими ангиогенных факторов (VEGF и др.). Последнее и объясняет, по-видимому, образование гемангиом и сильную васкуляризацию других VHL-ассоциированных опухолей, в частности светлоклеточных раков почки.
3.2.8. NFI/нейрофибромин и NР2/мерлин — гены нейрофиброматоза
Опухолевый супрессор NF1 коди рует белок нейрофибромин, состоя щий из 2818 аминокислот. В клетках, происходящих из нервного гребня, он регулирует активность онкобелков се мейства Ras, осуществляя перевод их функционально-активных ГТФ-свя- занных форм в неактивные ГДФ-свя- занные (см. раздел 3.1). Врожденные инактивирующие мутации в одном из аллелей гена NF1 (как правило, эти мутации прекращают синтез белка) вызывают развитие нейрофиброматоза I типа (болезнь Реклингхаузена) — на следственного заболевания, поражаю щего примерно 1 из 3500 индивидуу мов. Оно характеризуется возникнове нием опухолей периферической нерв ной системы, дефектами развития и нередко возникновением множествен ных новообразований различных ти пов в нескольких органах, в клетках которых действительно отмечается повышение активности онкобелков Ras. Мутации гена NF1 соответствуют
148
двухударной модели Кнудсона: в опу |
го торможения |
размножения |
клеток |
||||||||||||||||||
холевых |
клетках |
наблюдается пораже |
(см. раздел 2.1). При образовании |
||||||||||||||||||
ние и второго аллеля, что ведет к пол |
контактов клеток друг с другом и с |
||||||||||||||||||||
ному отсутствию в клетке белкового |
компонентами внеклеточного |
матрик- |
|||||||||||||||||||
продукта. У трансгенных мышей гете |
са происходит повышение экспрессии |
||||||||||||||||||||
розиготный нокаут этого гена (NF1+/—) |
мерлина, |
его дефосфорилирование и |
|||||||||||||||||||
вызывают |
|
не |
нейрофиброматоз, |
а |
переход |
в |
другую |
конформационную |
|||||||||||||
феохромоцитомы |
и миелоидные |
лей |
форму. В таком состоянии он переста |
||||||||||||||||||
козы — |
заболевания, |
наблюдающиеся |
ет связывать эзрин и моэзин, но оста |
||||||||||||||||||
у 1 % пациентов с нейрофибромато- |
ется |
связанным |
с |
внутриклеточным |
|||||||||||||||||
зом I типа. |
|
|
|
|
|
|
|
|
доменом |
рецептора |
гиалуроновой |
ки |
|||||||||
Врожденные |
мутации |
другого |
опу |
слоты (CD44), что ведет к блокирова |
|||||||||||||||||
холевого |
супрессора, |
NF2, |
вызывают |
нию |
проведения |
митогенного |
сигнала |
||||||||||||||
нейрофиброматоз II типа — наследст |
как от рецепторов ростовых факторов |
||||||||||||||||||||
венное заболевание, поражающее 1 из |
(вероятно, |
они ингибируются |
освобо |
||||||||||||||||||
40000 |
индивидуумов. |
|
Несмотря |
на |
дившимися белками ERM), так и от |
||||||||||||||||
название, при нем развиваются не |
CD44. Кроме CD44 и белков ERM, |
||||||||||||||||||||
нейрофибромы, а невриномы (шван- |
мерлин может также связывать бетаI-ин- |
||||||||||||||||||||
номы), менингиомы и эпендимомы. |
тегрин, бетаII-спектрин, H G S (субстрат |
||||||||||||||||||||
Наиболее |
характерным |
является |
воз |
тирозинкиназы, |
активируемой |
H G F / |
|||||||||||||||
никновение |
двусторонних |
неврином |
SF, — см. раздел 7.2) и др., но функ |
||||||||||||||||||
акустического нерва. NF2 ведет себя |
циональное |
значение этих |
взаимодей |
||||||||||||||||||
как классический |
опухолевый супрес |
ствий пока не выяснено. |
|
|
|
|
|
||||||||||||||
сор — его гомозиготные мутации, |
|
|
|
|
|
|
|
|
|
|
|
||||||||||
прекращающие |
синтез |
полноценного |
3.2.9. Ген WT1 и опухоль |
|
|||||||||||||||||
белка, наблюдаются не только при на |
|
||||||||||||||||||||
Вильмса |
|
|
|
|
|
|
|
||||||||||||||
следственных, но и при спорадиче |
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
ских невриномах и менингиомах. У |
Терминальные гетерозиготные |
му |
|||||||||||||||||||
трансгенных мышей с нокаутом одно |
тации гена |
WTl (Wilms' |
Tumor |
I) |
вы |
||||||||||||||||
го из аллелей гена NF2 резко увеличи |
зывают дефекты |
развития |
мочеполо |
||||||||||||||||||
вают вероятность развития во второй |
вой системы, в части случаев сопрово |
||||||||||||||||||||
половине |
жизни |
различных опухолей. |
ждающиеся |
возникновением |
|
в |
дет |
||||||||||||||
Их спектр сильно отличается от на |
ском |
возрасте нефробластом |
|
(опухо |
|||||||||||||||||
блюдаемого у пациентов с нейрофиб- |
лей Вильмса). Нарушения функции |
||||||||||||||||||||
роматозом II типа — образуются не |
гена |
WT1 |
ответственны |
за |
10—15 % |
||||||||||||||||
невриномы и менингиомы, а метаста- |
всех |
опухолей |
Вильмса |
(остальные |
|||||||||||||||||
зирующие |
|
остеосаркомы, |
фибросар- |
случаи связаны с изменениями других |
|||||||||||||||||
комы, аденокарциномы легкого и дру |
генов |
— |
WT2, |
WT3 и др., продукты |
|||||||||||||||||
гие новообразования. В то же время у |
которых не идентифицированы). Ген |
||||||||||||||||||||
мышей |
с |
гетерозиготными |
мутациями |
WT1 ведет себя как классический опу |
|||||||||||||||||
гена NF2, аналогичными тем, которые |
холевый супрессор, |
инактивирующий- |
|||||||||||||||||||
наблюдаются у пациентов с нейро- |
ся по двухударному механизму. Инак |
||||||||||||||||||||
фиброматозом |
II |
типа, |
развиваются |
тивация одного из аллелей гена |
WT1 у |
||||||||||||||||
невриномы. |
|
|
|
|
|
|
|
|
мышей также приводит к дефектам |
||||||||||||
Продукт гена NF2 — белок мерлин |
развития |
мочеполовой |
системы, |
но |
|||||||||||||||||
(другое название — шванномин) име |
частота возникновения |
опухолей |
при |
||||||||||||||||||
ет высокую степень гомологии с эзри- |
этом не повышается. |
|
|
|
|
|
|||||||||||||||
ном, радиксином и моэзином — пред |
Ген WT1 имеет очень сложную ре |
||||||||||||||||||||
ставителями |
семейства |
белков |
ERM, |
гуляцию. Его м Р Н К подвергается аль |
|||||||||||||||||
соединяющими |
|
интегральные |
мем |
тернативному сплайсингу, |
редактиро |
||||||||||||||||
бранные белки с актиновым цитоске- |
ванию и имеет несколько альтерна |
||||||||||||||||||||
летом. Мерлин играет, по-видимому, |
тивных |
сайтов |
инициации |
трансля |
|||||||||||||||||
важную роль в реализации контактно |
ции. |
В результате |
образуется |
24 |
изо- |
||||||||||||||||
149
формы белков WT1, многие из кото рых функционируют как транскрип ционные факторы, способные вызы вать как активацию, так и репрессию транскрипции генов. Пока достоверно идентифицирована лишь одна специ фическая мишень транскрипционной функции WT1: некоторые его продук
ты |
трaнс-активируют |
ген амфирегу- |
лина, |
представителя |
семейства E G F |
(факторов роста эпидермиса), регули рующего размножение и дифференцировку клеток. Связываясь с рецепто ром E G F (EGF - R или протоонкоген ErbBl — см. раздел 3.1), амфирегулин вызывает в одних типах клеток стиму ляцию размножения, а в других, в ча стности в эмбриональных эпителиаль ных клетках почки, — ингибирование пролиферации и индукцию специфи ческой дифференцировки. Кроме то го, некоторые изоформы белка WT1 связываются с р53, что может приво дить к изменению активности р53респонсивных генов. Механизмы кан церогенеза при нарушениях функции WT1 изучены плохо.
3.2.10. Мутаторные гены
Нарушения функций рассмотрен ных выше белков, контролирующих
апоптоз и/или клеточный цикл (р53, pRb, pl61NK4a, pARF и др.), отменяют
запрет на пролиферацию клеток с различными аномалиями, в том чис ле и с генетическими изменениями, что увеличивает вероятность появле ния онкогенных клеточных клонов. Эту группу белков принято называть gatekeepers — "сторожа". Наряду с этим идентифицирован ряд компо нентов специализированных систем распознавания и репарации повреж дений Д Н К , дисфункция которых также вызывает генетическую неста бильность, предопределяющую раз витие новообразований. Они получи ли название caretakers — "смотрите ли". Эта вторая группа белков и явля ется предметом рассмотрения данно го раздела.
В зависимости от типа поврежде
ний Д Н К могут активироваться три типа репарационных систем:
•системы репарации двунитевых разрывов Д Н К ;
•системы репарации неспаренных оснований (mismatch repair);
•системы эксцизионной репара ции.
Описаны наследственные формы новообразований, связанные с врож денными мутациями генов, продукты которых обеспечивают активацию и функционирование каждой из этих систем, причем некоторые из этих белков (ATM, СНК2, р53, BRCA1) ак тивируют также и молекулы, ответст венные за остановку клеточного цик ла и индукцию апоптоза, выполняя, таким образом, одновременно функ ции и "смотрителя", и "сторожа".
3.2.10.1. ATM, ATR, NBS1, СНК1 и СНК2 — компоненты систем проведения сигналов от поврежденной ДНК к различным эффекторам
Ключевую роль в интеграции сиг налов от поврежденной Д Н К и их дальнейшей передаче к разнообраз ным эффекторам играют специфиче ские протеинкиназы ATM (Ataxia-Tel angiectasia Mutated), ATR (ATM Relat ed), NBS1, CHK1 и CHK2 (чекпойнткиназы 1, 2) (схема 3.13). Белок ATM, имеющий структурное сходство с фос- фатидилинозит-3-киназой (PI3K), на капливается в местах повреждений и приобретает киназную активность, связывая фосфорилированные белки хроматина (Н2АХ и др.) и белки — сенсоры нарушений структуры ДНК. При этом ATM активируется в ответ на возникновение двунитевых разры вов Д Н К (вызываются гамма-облучением, ингибиторами топоизомераз и т. д.), тогда как другие нарушения структу ры Д Н К (например, сшивки основа ний, вызываемые УФ-облучением, или повреждения, индуцируемые алкилирующими соединениями) не ак тивируют ATM. В этих случаях, как и
150
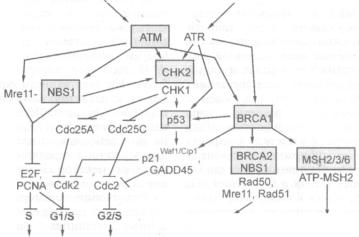
С х е м а 3.13. Сигнальные пути, регулирующие реакции клетки на
повреждения ДНК. Выделены компоненты, терминальные мута ции которых ответственны за наследственные синдромы, харак теризующиеся предрасположенностью к развитию определенных новообразований
гамма-Облучение |
УФ-облучение, |
|
блок синтеза ДНК |
||
|
|
Репарация |
Репарация |
Остановка клеточного цикла |
двунитевых |
неспаренных |
разрывов ДНК |
оснований |
при ингибировании синтеза Д Н К , на блюдается функциональная активация гомолога ATM, белка ATR. Активиро ванные формы ATM и ATR фосфорилируют ряд своих мишеней, в частно
сти р53 |
(см. |
раздел |
3.2.2), |
M r e l l , |
||
NBS1, |
С Н К 1 , |
С Н К 2 |
и BRCA1 (см. |
|||
схему |
3.2). |
|
|
|
|
|
Для фосфорилирования С Н К 2 не |
||||||
обходимо |
предварительное фосфори |
|||||
лирование |
белков комплекса |
M r e l l / |
||||
NBS1/Rad50, который, |
локализуясь |
в |
||||
местах |
повреждений, |
рекрутирует |
к |
|||
ним различные молекулы, в том числе СНК2, BRCA1, E2F и PCNA (привле чение PCNA вызывает переключение с репликативного синтеза Д Н К на ре парационный и остановку клеточного цикла в S-фазе; к блокированию вхо да и продвижения по S ведет и подав ление функции E2F — см. раздел 3.2.1.2). Фосфорилированные чекпойнткиназы СНК1/2 в свою очередь фосфорилируют и инактивируют бел ки семейства Cdc25, что вызывает по давление активности регулируемых
ими циклинзависимых киназ и быст рую остановку клеточного цикла в G1 (если Cdc25A не активирует cdk2) или в G2 (когда Cdc25C не активирует Cdc2). Кроме того, С Н К 1 и С Н К 2 ам-
плифицируют |
сигналы |
к |
р53 и |
||
BRCA1, |
что |
способствует длительной |
|||
задержке |
в |
G1 |
или G2 |
(см. |
разделы |
3.2.2.3 и 3.2.10.2) и, кроме того, акти визирует системы репарации Д Н К (см. схему 3.13 и разделы 3.2.10.3 и 3.2.10.4).
Терминальные инактивирующие мутации обоих аллелей гена ATM вы зывают атаксию-телеангиэктазию (AT) — тяжелое заболевание, характе ризующееся нейродегенерацией, им мунодефицитом и повышенным рис ком возникновения новообразова ний. Примерно у 10 % пациентов с AT в молодом возрасте развиваются лимфоидные опухоли из Т- или В- клеток (лимфосаркомы, лимфограну лематоз, различные формы лейкозов), а также рак молочной железы. Сома тические гомозиготные мутации гена
151
ATM характерны и для некоторых форм ненаследственных лимфолейкозов (Т-клеточного пролимфоцитарного лейкоза, В-клеточного хроническо го лимфолейкоза и др.). Гомозигот ный нокаут гена ATM у мышей также значительно увеличивает вероятность развития лимфоидных неоплазий. У индивидуумов с терминальными мута циями только одного из двух аллелей гена ATM несколько повышена часто та возникновения рака молочной же лезы. Онкогенный потенциал мутаций ATM связан, очевидно, с нарушения
ми реакций |
клетки |
на |
повреждения |
|
Д Н К и возникающей |
в |
связи с |
этим |
|
генетической |
нестабильностью. |
Так, |
||
после у-облучения в клетках с дефект ным ATM не происходит полноцен ной активации чекпойнтов и останов ки клеточного цикла в G1, S или G2. Кроме того, в них блокирована акти вация системы репарации двунитевых
разрывов Д Н К . |
В результате при |
инактивации ATM |
резко увеличивает |
ся вероятность размножения клеточ ных вариантов с различными генети ческими нарушениями.
Сходные последствия наблюдаются и при инактивации одной из важней ших мишеней ATM — белка NBS1. Терминальные гомозиготные мутации гена NBS1 вызывают ниймегенский синдром (Nijmegen Breakage Syn drome), характеризующийся Иммуно дефицитом, генетической нестабиль ностью и повышенной предрасполо женностью к развитию лимфоидных новообразований (в отличие от мута ций ATM мутации NBS1 не вызывают атаксию и телеангиэктазию). Сомати ческие мутации гена NBS1 выявляют ся в 10—20 % случаев ненаследствен ных форм острого лимфобластного лейкоза. В клетках с инактивацией
NBS1 |
наблюдается |
отмена остановки |
|
в S после у-облучения и понижение |
|||
эффективности работы систем |
репа |
||
рации |
двунитевых |
разрывов |
Д Н К |
вследствие нарушения функциониро вания комплекса Rad50/Mrel 1/NBS1, обеспечивающего оба механизма ис-, правления таких повреждений — го-
мологичную рекомбинацию ДНК и воссоединение концов разорванной Д Н К .
Потенциальным онкогенным эф фектом обладают, по-видимому, и на рушения функции белка ATR. Гетеро зиготный нокаут гена ATR у мышей приводит к увеличению частоты воз никновения лимфосарком, фибросарком, рака печени и яичника (инакти вация обоих аллелей гена ATR в отли
чие от гомозиготного |
нокаута |
гена |
|
ATM |
вызывает внутриутробную |
ги |
|
бель). |
Наследственного |
предрасполо |
|
жения к развитию каких-либо ново образований, связанного с врожден ными мутациями ATR, у людей пока не выявлено, но соматические мута ции этого гена нередко обнаруживают в клетках некоторых опухолей, в част ности рака желудка.
Увеличение риска развития ново образований наблюдается и при врож денных мутациях чекпойнткиназы СНК2 . Оказалось, что у части пациен тов с клиническими проявлениями синдрома Ли—Фраумени (см. раздел 3.2.2.1), но не имеющих мутаций р53, выявляют терминальные гетерозигот ные мутации гена СНК2. Этот факт свидетельствует о ключевой роли на рушений сигнального пути СНК2-р53, контролирующего реакции клетки на повреждения ДНК, в возникновении сильной предрасположенности к раз витию самых разных новообразова ний. Соматические инактивирующие мутации чекпойнткиназ СНК2 и СНК1 обнаруживаются в части случа ев наиболее распространенных опухо лей: рака легкого, толстой кишки, матки и др.
3.2.10.2. BRCA1 и BRCA2 контролируют репарацию ДНК и размножение клеток
Гены BRCA1 и BRCA2 были впер вые идентифицированы как гены, врожденные мутации которых ассо циированы с наследственными фор мами рака молочной железы. У жен щин с терминальными мутациями од-
152
ного из аллелей гена BRCA1 риск раз вития в течение жизни рака молочной железы составляет около 85 % (этот риск несколько варьирует в зависимо сти от местоположения и/или типа мутаций). Для опухолей яичника та кой риск несколько меньше — около 50 %. У носителей врожденных мута ций гена BRCA1 выше также вероят ность развития опухолей толстой кишки и предстательной железы. При терминальных мутациях гена BRCA2 риск развития опухолей молочной же лезы несколько ниже, чем при мута циях BRCA1. Отличительными черта ми мутаций BRCA2 являются более частое возникновение рака молочной железы у мужчин и меньший риск развития опухолей яичника. Гены BRCAJ и BRCA2 ведут себя как клас сические опухолевые супрессоры: для инициации опухолевого роста, поми мо врожденной мутации в одном из аллелей, необходима и инактивация второго аллеля, которая происходит уже в соматической клетке. Как пра вило, мутации в генах BRCA1 и BRCA2 ведут к прекращению синтеза полно размерного белка. Особенностью му таций генов BRCA1 и BRCA2 является то, что они характерны для наследст венных форм новообразований и зна чительно реже обнаруживаются в не наследственных опухолях той же ло кализации.
Гены BRCA1 и BRCA2 кодируют ядерные фосфобелки (соответственно 1863 и 3495 аминокислот), которые за счет разнообразных белок-белковых взаимодействий участвуют в регуля ции репарации Д Н К и размножения (клеток. Так, белок BRCA1 связывает белки, ответственные за гомологич ную рекомбинацию и репарацию дву нитевых разрывов Д Н К (Rad50, Rad51, BRCA2), компоненты систем
репарации неспаренных |
оснований |
Д Н К (MSH2, MSH6, MLH1, АТР- |
|
MSH2 и др.), транскрипционные фак |
|
торы (базальные - HDAC, |
рЗОО/СВР, |
SWI/SNF и сиквенсспецифические — Р53, MYC, E2F, ZBRK1, ATF, рецеп тор эстрогенов, рецептор андрогенов),
а также ряд других белков — pRb (см. раздел 3.2.1), BARD1 (опосредует убиквитинирование), ВАР1 (ответствен за деубиквитинирование), Nm23 (компо нент центросомы) и т. д. Транскрип ционная функция BRCA1 заключается в его способности репрессировать од ни сиквенсспецифические факторы транскрипции (MYC, E2F, рецептор эстрогенов и др.) и активировать дру гие (р53 и др.), модулируя, таким об разом, активность генов, регулируе мых этими факторами. При генотоксических стрессах (у-облучение и др.) транскрипционная функция BRCA1 направлена на индукцию остановки клеточного цикла по нескольким ме ханизмам. Так, она обеспечивает уси ление активности р53; включение дуб лирующих, р53-независимых, путей
активации некоторых р53-респонсив- ных генов (p21Wan/Cipl, GADD45), вы
зывающих задержку соответственно в G1 и G2 (см. раздел 3.2.2.3); подавле ние активности MYC (см. раздел 3.1), E2F (см. раздел 3.1.2.2) и т . д . Одно временно активированный BRCA1, взаимодействуя с белками репараци онных систем, стимулирует восста новление нормальной структуры Д Н К . Рекрутируя комплексы Rad50/ M r e l l / N B S l , он стимулирует процессирование концов разорванной Д Н К , подготавливая их либо для гомологич ной рекомбинации, либо для воссо единения конец в конец — двух ос
новных |
путей репарации двунитевых |
|
разрывов |
Д Н К . Взаимодействуя с |
|
комплексом Rad51/BRCA2, он |
увели |
|
чивает эффективность процесса |
гомо |
|
логичной рекомбинации Д Н К . Связы ваясь с белками MSH2, MSH3, MSH6 и др., BRCA1 участвует, очевидно, также и в работе системы репарации
неспаренных |
оснований |
(исправляет |
||
ошибки |
репликации |
Д Н К и непра |
||
вильную |
репарацию |
двунитевых раз |
||
рывов Д Н К — см. раздел 3.2.10.3). |
||||
Помимо |
контроля |
повреждений |
||
Д Н К и |
поддержания |
целости генома, |
||
BRCA1 |
выполняет и ряд других функ |
|||
ций. Так, он связывает рецептор эст рогенов и репрессирует его транс-
153
крипционную функцию, сдерживая, таким образом, избыточную пролифе рацию клеток молочной железы и других эстрогензависимых органов, в частности при половом созревании и беременности. Кроме того, BRCA1, взаимодействуя с компонентами цен тросом (Nm23 и др.), принимает уча стие в обеспечении правильной сегре гации хромосом во время митоза.
С учетом столь многочисленных функций BRCA1 становятся понятны ми последствия его инактивации. В клетках с дефектным BRCA1 наблю дается сильная генетическая неста бильность, т. е. повышение частоты возникновения спонтанных или инду цированных мутагенами генетиче ских изменений — генных мутаций, хромосомных транслокаций, анеуплоидии и т. д. Кроме того, отменяется сдерживание пролиферации эстрогензависимых клеток, что и объясняет, очевидно, возникновение опухолей именно молочной железы и яичника.
Функции белка BRCA2 изучены хуже. Как и BRCA1, он обладает репа рационной и транскрипционной ак тивностью. Связывая Rad51 (гомолог бактериального белка RecA), BRCA2 увеличивает его способность катали зировать рекомбинации Д Н К , обеспе чивающие репарацию двунитевых раз
рывов |
Д Н К . |
Транскрипционная |
|
функция BRCA2 |
связана, |
очевидно, |
|
со способностью |
рекрутировать Р/ |
||
CAF |
(р300/СВР |
Associated |
Factors), |
ацетилирующие гистоны и ремоделирующие хроматин, однако физиологи ческие гены-мишени BRCA2 пока не идентифицированы. Тем не менее о важности транскрипционной актив ности BRCA2 для его супрессорной функции может свидетельствовать тот факт, что обнаруживаемые в опухолях молочной железы мутации поражают именно транскрипционный домен. У мышей гомозиготный нокаут резко уменьшает жизнеспособность эмбрио нов, а у выживших животных развива ются злокачественные тимомы. На клеточном уровне инактивация BRCA2 приводит к гиперчувствитель
ности к различным генотоксическим агентам (УФ- и у-облучение, химиче ские мутагены), повышению частоты встречаемости незарепарированных двунитевых разрывов Д Н К и различ ных перестроек хромосом. Механиз мы специфического возникновения у пациентов с терминальными мутация ми BRCA2 опухолей молочной желе зы, яичника и предстательной железы пока не установлены.
3.2.10.3. MSH2, MSH6, MLH1 и PMS2 — компоненты систем репарации неспаренных оснований ДНК
Риск развития новообразований значительно повышается и при врож денных дефектах системы репарации неспаренных оснований (mismatch re pair), исправляющей главным образом ошибки репликации Д Н К и неточно сти репарации двунитевых разрывов. В результате таких ошибок и потери
комплементарности нитей ДНК |
воз |
||
никают петли, |
которые |
распознаются |
|
комплексами |
белков |
MSH2/MSH6 |
|
или MSH2/MSH3 (они отличаются по |
|||
способности |
узнавать |
разные |
типы |
петель, образующиеся при замене ос нований, инсерциях и делециях). Эти комплексы рекрутируют к местам с нарушенной структурой Д Н К ком плексы белков MLH1/PMS2 или MLH1/MLH3, которые в свою оче редь привлекают экзо- и эндонуклеазы, осуществляющие эксцизию ано мального фрагмента ДНК, а также факторы репликации (PCNA, ДНКполимеразы), обеспечивающие за стройку бреши и восстановление нор мальной структуры Д Н К .
Врожденные гетерозиготные мута ции по меньшей мере четырех из ком понентов этой системы — MSH2, MLH1, MSH6 и PMS2 - вызывают синдром Линча. Главной чертой этого синдрома является развитие в моло дом возрасте опухолей толстой кишки (так называемый наследственный неполипозный колоректальный рак) и/ или опухолей яичника. Преимущест-
154
венное возникновение |
опухолей |
ки |
эмбриогенезе клеток всех тканей не |
|||||||||||
шечника, вероятно, связано с высо |
обходимое |
для |
образования |
опухоли |
||||||||||
чайшим |
пролиферативным |
потенциа |
количество |
мутаций успевает |
нако |
|||||||||
лом клеток на дне кишечных крипт, |
питься в каких-то клетках задолго до |
|||||||||||||
что, естественно, ведет и к более час |
рождения, тогда как при гетерозигот |
|||||||||||||
тому |
появлению |
ошибок репликации, |
ных мутациях темп мутирования ниже |
|||||||||||
которые |
должны |
|
исправляться имен |
и накопление мутаций до критическо |
||||||||||
но системами репарации неспаренных |
го уровня продолжается в интенсивно |
|||||||||||||
оснований. Естественно, что бурно |
размножающихся клетках |
взрослого |
||||||||||||
размножающиеся |
|
полустволовые |
(ам- |
организма. С этой точки зрения пока |
||||||||||
плифицирующие) |
|
клетки |
кишечного |
непонятно, почему у мышей как с ге |
||||||||||
эпителия |
|
накапливают |
необходимый |
терозиготным, так и с гомозиготным |
||||||||||
для развития опухолей набор мутаций |
нокаутом гена MSH2 или гена MLH1 |
|||||||||||||
быстрее, |
чем медленно |
размножаю |
также развиваются лимфомы и сарко |
|||||||||||
щиеся клетки. |
|
|
|
|
|
|
мы, а не опухоли кишечника. (Впро |
|||||||
Возникновение |
опухолей, при |
дис |
чем, следует заметить, что мыши |
|||||||||||
функции |
|
MSH2, |
|
MLH1, |
|
MSH3 |
или |
сильно отличаются от человека и по |
||||||
PMS2 связано, очевидно, с повышен |
типу спонтанно |
развивающихся |
опу |
|||||||||||
ной вероятностью мутаций в протоон- |
холей: у человека большую часть но |
|||||||||||||
когенах |
и |
опухолевых |
супрессорах. |
вообразований |
представляют |
различ |
||||||||
Действительно, |
при |
мутациях |
гена |
ные формы рака, возникающие из |
||||||||||
MSH2 |
или |
MLH1 |
частота |
точечных |
эпителиоцитов, тогда как у мышей та |
|||||||||
мутаций во всех локусах увеличивает |
кие опухоли достаточно редки, а воз |
|||||||||||||
ся на 1—2 порядка, а в наследствен |
никают, как правило, лимфомы и сар |
|||||||||||||
ных колоректальных раках, как пра |
комы.) Природу таких различий еще |
|||||||||||||
вило, |
обнаруживаются |
точечные |
мута |
предстоит |
выяснить. |
|
|
|||||||
ции в генах р-катенина, АРС, TpR-II, |
|
|
|
|
|
|
|
|||||||
Smad2, Smad4 и т. д., которые, по-ви |
3.2.10.4. Компоненты системы |
|||||||||||||
димому, |
и являются |
причиной разви |
||||||||||||
эксцизионной репарации ДНК |
||||||||||||||
тия |
новообразований. |
Маркером |
||||||||||||
и пигментная ксеродерма |
||||||||||||||
инактивации любого из генов репара |
||||||||||||||
|
|
|
|
|
|
|
||||||||
ции |
неспаренных оснований |
служит |
Система |
эксцизионной реларации |
||||||||||
легко |
выявляемая |
нестабильность |
узнает и |
исправляеттлцивки |
основа |
|||||||||
микросателлитных |
последовательно |
ний (тиминовые димеры и др.), обра- |
||||||||||||
стей ДНК . Нарушения функции генов |
зующисся, например, после УФ-облу- |
|||||||||||||
MSH2, MLH1, MSH3, MSH6 и PMS2, |
чения или оксидативного стресса. Она |
|||||||||||||
приводящие к нестабильности микро |
включает |
множество |
|
компонентов. |
||||||||||
сателлитов, характерны и для некото |
Распознавание |
тиминовых |
димеров |
|||||||||||
рых форм спорадических |
(ненаследст |
осуществляется |
белковым комплексом |
|||||||||||
венных) |
опухолей: |
они |
обнаружива |
XPC-hHR23. который вызывает рек |
||||||||||
ются |
в |
13—15 % |
опухолей |
толстой |
рутирование |
к |
месту |
повреждения |
||||||
кишки, рака желудка и эндометрия, |
фактора ТFIIH -- сложного белкового |
|||||||||||||
но значительно реже (< 2 %) — в дру |
комплекса, состоящего из 9 субъеди |
|||||||||||||
гих новообразованиях. |
|
|
ниц и обладающего разными активно |
|||||||||||
Описаны единичные случаи терми |
стями, в том числе хеликазной и |
|||||||||||||
нальных мутаций обоих аллелей гена |
транскрипционной. |
|
Привлеченный |
|||||||||||
MLH1, которые приводили к разви |
фактор TFIIH катализирует раскрытие |
|||||||||||||
тию еще во внутриутробном возрасте |
поврежденного |
участка |
Д Н К |
и спо |
||||||||||
лимфосарком, |
лейкозов |
и нейрофиб |
собствует |
|
сборке |
репарационного |
||||||||
роматоза. |
Это |
объясняется, |
видимо, |
комплекса. Затем к дефектному участ |
||||||||||
тем, что при полной инактивации |
ку последовательно |
|
рекрутируются |
|||||||||||
системы |
репарации |
ошибок реплика |
белки ХРG, ХРА, комддекc RРА и, |
|||||||||||
ции |
Д Н К и бурном размножении в |
наконец, |
белки |
XPF - ERCC1, |
являю- |
|||||||||
155
щиеся эндонуклеазами. Именно они и осуществляют эксцизию поврежден ного участка Д Н К (обычно вырезается 24—32 нуклеотида) и инициируют за стройку бреши по неповрежденной матрице и восстановление нормаль ной структуры ДНК .
Терминальные гетерозиготные мутации компонентов системы эксцизионной репарации, в частности генов
ХРА, ХРВ, XPC, XPD, XPF, XPG, ведут
квозникновению пигментной ксеродермы — наследственного заболева ния, характеризующегося повышен ной чувствительностью к ультрафио летовому облучению и развитием множественных опухолей кожи на местах, подвергающихся солнечному облучению. Интересно, что, несмотря на участие эксцизионной репарации в исправлении дефектов, вызванных не только УФ-облучением, но и мутаге нами/канцерогенами, частота возник новения других форм опухолей при пигментной ксеродерме почти не уве личивается. При этом у трансгенных мышей с аналогичными дефектами системы эксцизионной репарации от мечается повышение частоты индук ции новообразований химическими канцерогенами. Преимущественное возникновение у пациентов с пиг ментной ксеродермой исключительно опухолей кожи может указывать на незначительную роль химических факторов, загрязняющих окружаю щую среду, в развитии опухолей внут ренних органов у человека.
** *
Кнастоящему времени идентифи цировано несколько десятков генов, инактивация которых приводит к раз витию новообразований. Большинст во из них, регулируя клеточный цикл, апоптоз или репарацию Д Н К , предот вращают накопление в организме кле ток с генетическими и некоторыми другими аномалиями. Выявлены опу холевые супрессоры и с другими функциями, в частности контролирующие морфогенетические реакции
клетки и ангиогенез. Обнаруженные гены далеко не исчерпывают список существующих опухолевых супрессо ров. Уже сейчас в хромосомах челове ка картировано более сотни участков, закономерно делегирующихся при различных новообразованиях и, сле довательно, содержащих потенциаль ные опухолевые супрессоры. Их иден тификация, вероятно, приведет к об наружению и других путей подавле ния опухолевого роста. В ближайшем будущем можно ожидать также успехов в прикладном использовании знаний об опухолевых супрессорах. Следует надеяться, во-первых, на разработку высокоточных методов диагностики наследственных синдромов, предрас полагающих к развитию новообразо ваний (см. раздел 12.2), а во-вторых, на создание принципиально новых методов терапии злокачественных но вообразований, основанных на моди фикации сигнальных путей, контро лируемых опухолевыми супрессорами.
Рекомендуемая литература
Копит Б. П. Мишени действия онкогенов и опухолевых супрессоров: ключ к по ниманию базовых механизмов канце рогенеза // Биохимия. — 2000. — № 65. - С. 5 - 3 3 .
Чумаков П. М. Функция гена р53: выбор между жизнью и смертью // Биохи мия. - 2000. - № 65. - С. 34-47.
Bienz М., Clevers Н. Linking colorectal cancer to Wnt signaling // Cell. - 2000. - Vol. 103. - P. 311-320.
Blagosklonny M. V. Do VHL and HIF-1 mir ror p53 and Mdm-2. Degradation-transac- tivation loops of oncoproteins and tumor suppressors // Oncogene. — 2001. —
Vol. 20. - P. 395-398. |
|
|||
Bonneau D., Longy M. Mutations |
of the hu |
|||
man PTEN gene // Hum. |
Mutat. — |
|||
2000. - Vol. 16. - P. 109-122. |
||||
tie Laat W. L., Jaspers |
N. G. J., |
Hoeijmakers |
||
J. H. J. |
Molecular |
mechanism |
of nucleo |
|
tide excision repair // Gen. Dev. — 1999. — |
||||
Vol. 13. - P. 768785 . |
|
|||
Ghebranious |
N, Donehower L. A. Mouse mod |
|||
els in tumor suppression // Oncogene. — |
||||
1998. - Vol. 17. - P. 3385-3400. |
||||
Grana X., |
Garriga |
J., |
Mayol X. Role of the |
|
retinoblastoma |
protein family, |
pRb, pl07 |
||
156
and pi30 in the negative control of cell growth // Oncogene. — 1998. — Vol. 17.
-P. 3365-3383.
Gray J. W., Collins C. Genome changes and gene expression in human solid tumors // Carcinogenesis. — 2000. — Vol. 21. — P. 443-452.
Gutmann D. H., Hirbe A. C, Haipek C. A.
Functiomal analysis of neurofibromatosis 2 (NF2) missense mutations // Hum. Mol. Genet. - 2001. — Vol. 10. — P. 1519-1529.
Hanahan D., Weinberg R. A. The hallmarks of cancer // Cell. — 2000. — Vol. 100. — P. 57 - 70 .
Harbour/. W., Dean D. C. The Rb/E2F path way: expanding roles and emerging para digms // Genes Dev. — 2000. — Vol. 14.
-P. 2393-2409.
Hooper M. L. Tumour suppressor gene muta tions in humans and mice: parallels and contrasts // EMBO J. — 1998. - Vol. 17.
-P. 6783-6789.
Kastan M. В., Lim D. S. The many substrates and functions of ATM // Nature Rev. Mol. Cell. Biol. - 2000. - Vol. 1. - P. 179-186.
Kinzler K. W., Vogelstein B. Gatekeepers and caretakers // Nature. — 1997. — Vol. 386.
-P. 761 - 763 .
Knudson A. G. Mutation and cancer: statistical study of retinoblastoma // Proc. Natl. Acad. Sci. USA. - 1971. - Vol. 68. - P. 820 - 823 .
Kolodner R. D., Marsischky G. T. Eukaryotic DNA mismatch repair // Curr. Opin. Genet. Dev. - 1999. — Vol. 9. - P. 8 9 - 96.
Lehman A. R. The xeroderma pigmentosum group D (XPD) gene: one gene, two func tions, three diseases // Gen. Dev. — 2001.
-Vol. 15. — P. 15 - 23 .
Lengauer C, Kinzler K. W., Vogelstein B. Ge netic instabilities in human cancers // Na ture. - 1998. - Vol. 396. - P. 643-649.
Levine A. J. p53, the celular gatekeeper for growth and division // Cell. — 1997. — Vol. 88. - P. 323 - 331 .
Levine A. J. The tumour suppressor genes // Annu. Rev. Biochem.. — 1993. — Vol. 62.
-P. 623 - 651 .
Lipinski M. M., Jacks T. The retinoblastoma gene family in differentiation and develop ment // Oncogene. — 1999. — Vol. 18. — P. 7873-7882.
Maehama Т., Dixon J. E. PTEN: a tumour suppressor that functions as a phospholipid phosphatase // Trends Cell Biol. — 1999. - Vol. 9. - P. 125-128.
Massague J. |
How cells read TGF - p signals |
// |
Nature Rev. Mol. Cell Biol. — 2000. |
— |
|
Vol. 1. - P. 169-178. |
|
|
Massague J., |
Blain S. W., Lo R. S. TGFb sig |
|
naling in growth control, cancer, and her |
||
itable |
disorders // Cell. — 2000. |
— |
Vol. 103. - P. 295-309. |
|
|
Paggi M. G, Giordano A. Who is the boss |
in |
|
the retinoblastoma family? The point of view of Rb2/pl30, the little brother // Cancer Res. — 2001. — Vol.61. — P. 4651-4654.
Polakis P. More than one way to skin a catenin // Cell. - 2001. - Vol. 105. - P. 563-566.
Polakis P. The adenomatous polyposis coli (APC) tumor suppressor // Biochim. Biophys. Acta. - 1997. - Vol. 1332. - P. F127 - F147 .
Polakis P. The oncogenic activation of p-cat- enin // Curr. Opin. Genet. Dev. — 1999.
-Vol. 9. - P. 1 5 - 2 1 .
Polakis P. Wnt Signaling and cancer // Genes Dev. - 2000. - Vol. 14. - P. 1837-1851.
Ponder B. A. J. Cancer genetics // Nature. —
2001. |
- Vol. 411. - P. 336 - 341 . |
|
|
Prives C, |
Hall P. A. |
The p53 pathway |
// J. |
Path. |
- 1999. |
- Vol. 187. - P. |
1 1 2 - |
126. |
|
|
|
Ruas M., Peters G. The pl61NK4a/CDKN2A tumor suppressor and its relatives // Bio chem. Biophys. Acta. — 1998. — Vol. 1378. - P. F115 - F177 .
Sherr C. J. Parsing Ink4a/Arf: "pure" pl6-null
mice // Cell. |
- 2001. - |
Vol. |
106. - |
||
P. 531-534. |
|
|
|
||
Sherr |
C. J., |
Weber J. D. The ARF/p53 path |
|||
way // Curr. Opin. Genet. Dev. — 2000. |
|||||
- |
Vol. |
10. - P. |
9 4 - 9 9 . |
|
|
Shiloh |
Y. |
Ataxia-telangiectasia |
and |
the Ni- |
|
jmegen breakage syndrome: related disor ders but genes apart // Annu. Rev. Genet.
-1997. - Vol. 31. - P. 635-662.
Sionov R. |
V., Haupt Y. |
The cellular response |
|||||
to p53: the decision between life |
and |
||||||
death // Oncogene. — 1999. — Vol. 18. — |
|||||||
P. 6145-6157. |
|
|
|
|
|
||
Taipale J., |
Beachy |
P. A. The Hedgehog and |
|||||
Wnt signaling pathways in cancer // Na |
|||||||
ture. |
- |
2001. |
- Vol. 411. |
- |
P. 3 5 0 3 - |
||
3540. |
|
|
|
|
|
|
|
The Genetic Basis of Human Cancer / Eds B. |
|||||||
Vogelstein, K. W. Kinzler. — |
New York: |
||||||
McGraw Hill, |
1998. |
|
|
|
|
||
Vogelstein |
В., Lane |
D., |
Levine A. |
Surfing |
the |
||
p53 |
network |
// ' Nature. |
— |
2000. |
— |
||
Vol. 408. - P. 307-310.
Walworth N. C. Cell-cycle checkpoint kinases: checking in on the cell cycle // Curr.
157
Opin. |
|
Cell |
Biol. |
- 2000. |
- Vol. |
12. |
- |
цитокинам необходимо отнести и зна |
|||||||||||||||
P. 697-704. |
|
|
|
|
|
|
|
чительное |
число |
мембраносвязанных |
|||||||||||||
Wang Q., |
|
Zhang |
PL., |
Fishel |
R., |
Greene |
M. I. |
молекул, |
которые |
действуют |
на ре |
||||||||||||
BRCA1 |
|
and cell signaling // Oncogene. — |
цепторы по тому же механизму, что и |
||||||||||||||||||||
2000. |
- |
|
Vol. |
19. |
- P. 6152-6158. |
|
|
||||||||||||||||
|
|
|
растворимые |
цитокины, но |
требуют |
||||||||||||||||||
Weinberg |
R. A. |
The |
molecular basis of onco |
||||||||||||||||||||
непосредственного |
контакта |
между |
|||||||||||||||||||||
genes and |
tumor suppressor genes // Ann. |
||||||||||||||||||||||
клетками. |
|
|
|
|
|
|
|
|
|||||||||||||||
N. Y. |
Acad. |
Sci. |
- 1995. |
- Vol. |
758. |
- |
|
|
|
|
|
|
|
|
|||||||||
Тот факт, что цитокины способны |
|||||||||||||||||||||||
p 231 |
|
|
338 |
|
|
|
|
|
|
|
|||||||||||||
Welsh P. A., King M.-C. BRCA1 and BRCA2 |
передавать |
|
сигналы |
|
пролиферации, |
||||||||||||||||||
and the genetics of breast and ovarian can |
дифференцировки |
и |
апоптоза, сразу |
||||||||||||||||||||
cer // Hum. Mol. Genet. - 2001. — |
позволяет |
предположить |
существен |
||||||||||||||||||||
Vol. 10. - P. 705 - 713 . |
|
|
|
|
ную роль цитокинов, |
их рецепторов и |
|||||||||||||||||
Woods D. |
В., |
Vousden К. H. Regulation of p53 |
внутриклеточных |
сигнальных |
меха |
||||||||||||||||||
function // Exp. Cell Res. — 2001. - |
|||||||||||||||||||||||
низмов |
в |
|
процессах, |
связанных с |
|||||||||||||||||||
Vol. |
264. - P. |
56 - 66 . |
|
|
|
|
трансформацией |
клеток. |
Имеются |
||||||||||||||
Yang A., |
McKeon |
F. |
p63 and |
p73: p53 |
mim |
||||||||||||||||||
примеры |
неоплазий, |
|
вызванных по |
||||||||||||||||||||
ics, |
menaces and |
more // Nat. Rev. Mol. |
|
||||||||||||||||||||
стоянной |
активацией |
или |
инактива |
||||||||||||||||||||
Cell. |
Biol. - |
2000. - Vol. 1. — P. |
199— |
||||||||||||||||||||
цией конкретных |
сигнальных |
цепей. |
|||||||||||||||||||||
207. |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
В то же время недавно было установ |
|||||||||||
Zheng L., |
Li S., Boyer T. G., Lee W.-H. Les |
||||||||||||||||||||||
sons learned from BRCA1 and BRCA2 // |
лено, что активность некоторых цито |
||||||||||||||||||||||
Oncogene. |
|
— |
|
2000. |
— |
Vol. 19. |
— |
кинов |
используется |
организмом при |
|||||||||||||
P. 6159-6175. |
|
|
|
|
|
|
осуществлении |
|
противоопухолевого |
||||||||||||||
Zhou B.-B. S., Elledge S. J. The DNA damage |
надзора. Наконец, биологические ак |
||||||||||||||||||||||
response: putting checkpoints in perspec |
тивности многих цитокинов как in vit |
||||||||||||||||||||||
tive |
// |
|
Nature. |
- 2000. |
- Vol. |
408. |
- |
||||||||||||||||
P. 433-439. |
|
|
|
|
|
|
|
ro, так |
и in |
vivo |
могут быть использо |
||||||||||||
|
|
|
|
|
|
|
ваны в клинической практике, что де |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
3.3. Цитокины, их рецепторы |
|
лает приобретение знаний в этой бы |
|||||||||||||||||||||
|
стро развивающейся области биологи |
||||||||||||||||||||||
и передача внутриклеточных |
ческой |
науки |
особенно |
актуальным |
|||||||||||||||||||
сигналов |
|
|
|
|
|
|
|
при подготовке врачей и клинических |
|||||||||||||||
С. А. |
|
Недоспасов, Д. |
В. |
Купраш |
|
исследователей. |
|
|
|
|
|
|
|||||||||||
Цитокинами первоначально были названы секретируемые белковые (по липептидные) медиаторы передачи сигналов между клетками, действую щие через специфические рецепторы. К таким сигналам относятся сигналы активации, пролиферации, дифферен цировки, программируемой клеточ ной смерти и некоторые другие. Меж ду растворимыми цитокинами, дейст вующими системно и дистально, и не которыми гормонами (которые проду цируются специализированными же лезами и также действуют дистально) существует сходство, так что некото рые из них, например гормон роста или эритропоэтин, могут быть класси фицированы и как гормоны, и как цитокины. Позже, когда механизмы передачи сигналов цитокинов были подробно изучены, стало ясно, что к
3.3.1. Классификация цитокинов и их рецепторов
Наиболее логичная классификация цитокинов основана на способе пере дачи внутриклеточного сигнала, т. е. на структуре не самих цитокинов, а их рецепторов. Согласно классификации, выделяют четыре главных семейства.
1. Рецепторы, которые в своих внутриклеточных участках содержат "встроенные" киназы — тирозиновые (некоторые рецепторы гемопоэтических факторов и других факторов рос та) или серин-треониновые (семейст во трансформирующего фактора рос та — бета). Мутантные формы неко торых рецепторов этого типа являют ся клеточными онкогенами (табл. 3.6).
2. Рецепторы не содержат "встро енных" киназных доменов, но в ответ
158
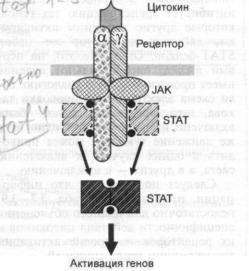
Т а б л и ц а 3.6. Рецепторы некоторых ци-
токинов, содержащие "встроенные" киназы
Ли- |
Рецептор(ы) |
|
Тип киназы |
|
ганд(ы) |
|
|||
|
|
|
|
|
|
|
|
|
|
E G F |
E G F R |
|
|
Тирозиновая |
P D G F |
P D G F R |
|
" |
|
VEGF |
VEGFR - 1 |
(Flt-1) |
" |
|
|
VEGFR - 2 |
|
|
|
|
(KDR/Flk-1) |
|
|
|
|
V E G F R3 |
(Flt-4) |
|
|
TGFбета |
TGFбетаR-I |
|
|
Серинтрео- |
|
TGFбетаR-II |
|
|
ниновая |
CSF-1 |
M - CSFR |
|
|
Тирозиновая |
(M-CSF) |
|
|
|
|
SCF |
c-Kit |
|
|
" |
|
|
|
|
|
|
|
|
|
|
на связывание цитокина способны олигомеризоваться и рекрутировать в рецепторный комплекс киназы семей ства JAK (состоящего из JAK1-3 и TYK2), которые и запускают внутри клеточный сигнал (схема 3.14). Отли чительной особенностью этого класса рецепторов и цитокинов (к ним отно сится большинство интерлейкинов, интерферонов, гормон роста и неко торые гемопоэтины) является наличие стадии фосфорилирования транс крипционных факторов семейства STAT, которые также привлекаются в рецепторный комплекс в цитоплазме и после фосфорилирования транслоцируются в ядро (табл. 3.7). По сути дела в результате фосфорилирования JAK и/или рецепторов возникает стерическая возможность пристыковки к рецепторному комплексу неактивных транскрипционных факторов, кото рые впоследствии активируются.
Как видно из табл. 3.7, за исключе нием ИЛ-12, ни один из рецепторов не передает уникального сигнала на транскрипционные факторы. Это оз начает, что специфичность сигнала, приводящего к активации характерно го набора генов, объясняется не толь ко типом транскрипционного фактора (и соответствующего участка связыва ния в регуляторном элементе гена), но и дополнительными параметрами,
такими как кинетика активации, сила, сигнала активации (связанная, напри мер, с количеством рецепторов на клетках), наличие и концентрация ре-
гуляторных и сигнальных |
молекул, |
что определяется клеточным |
контек |
стом — видом клетки, стадией дифференцировки и т. д. Тем не менее ли митирующей стадией для активации генов-мишеней может быть единст венный транскрипционный фактор, который включит только те гены, для которых все другие условия, необхо димые для их активации, выполнены. Это можно легко пояснить на приме ре системного тумблера, который при включении, во-первых, приведет в действие только те приборы, у кото рых в активном положении находится еще один выключатель, и, во-вторых, может включить или выключить со вершенно разные приборы, функции которых могут быть совершенно не
схожи и даже противоположны. |
|
|
Рецепторы ИЛ-10, |
действуя |
через |
те же JАК-киназы и STAT-белки, что |
||
С х е м а 3.14. Передача |
сигнала |
через |
нитокиновые рецепторы, |
приведенные в |
|
табл. 3.7. Эти рецепторы могут иметь вид не только гетеродимеров, но и гомодимеров или гетеротримеров
159
Т а б л и ц а 3.7. Рецепторы некоторых цитокинов, ассоциированных с JAK киназами
|
|
|
|
|
Активи |
Субъедини |
|
|
|
|
|
Ассоцииро |
ца, общая с |
||
|
|
|
|
руемые |
|||
Лиганд |
Субъединицы рецептора |
ванные кина |
другими |
||||
STAT- |
|||||||
|
|
|
|
зы |
рецепто |
||
|
|
|
|
белки |
|||
|
|
|
|
|
рами |
||
|
|
|
|
|
|
||
|
|
|
|
|
|||
ИЛ-2 |
CD25, CD122, CD132(γc) |
JAK1, 3 |
1,3, 5a/b |
γс |
|||
ИЛ-3 |
CD123, |
CD13I(βc) |
JAK2, TYK2 |
1,3, 5a/b |
βс |
||
ИЛ-4 |
CD 124, CD132(γc) |
JAK1, 3 |
5a/b, 6 |
γс |
|||
ИЛ-5 |
CD125, |
CD131(βc) |
JAK1, 2 |
1, 5a/b |
βс |
||
ИЛ-6 |
CD126, |
CD1302(gpl30) |
JAK1, 2; TYK2 |
1,2, 3 |
gpl30 |
||
ИЛ-7 |
CD 127, CD132(γc) |
JAK1, 3 |
3, 5a/b |
γс |
|||
ИЛ-9 |
IL9R, CD132(γc) |
JAK1, 3; TYK2 |
3, 5a/b |
γс |
|||
ИЛ-10 |
IL10R α, |
β |
JAK1; TYK2 |
1,3 |
— |
||
ИЛ-11 |
IL11R, |
CD130(gpl30) |
JAK1, 2; TYK2 |
1,2,3 |
gpl30 |
||
ИЛ-12 |
1L12R, CD212 |
JAK2; TYK2 |
4 |
Рецептор |
|||
|
|
|
|
|
|
похож на |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
gpl30 |
|
ИЛ-13 |
CD124, |
CD213 |
JAK1,3; TYK2 |
3, 5a/b, 6 |
γс |
||
ИЛ-15 |
1L15R, CD122, CD132(γc) |
JAK1, JAK3 |
3, 5a/b |
γс |
|||
ИЛ-21 |
IL21R, |
CD132(γc) |
JAK1, JAK3 |
1, 3 |
γс |
||
ИФН - α / β |
CD1182 |
|
|
JAK1, 3; TYK2 |
1,2,3, 5a/b |
— |
|
ИФН - γ |
CD1192 |
|
|
JAK1, 2 |
1,3, 5a/b |
— |
|
Г-КСФ |
CD114 |
|
|
JAK 1,2; TYK2 |
1,3, 5a/b |
— |
|
ГМ - КСФ |
CD116, CD131(βc) |
JAK2, TYK2 |
1,3, 5a/b |
βс |
|||
ЭПО |
EpoR |
|
|
JAK.2 |
5a/b |
— |
|
Гормон роста (ГР) |
Рецептор |
ГР |
JAK1,2 |
1,3, 5a/b |
— |
||
Пролактин (П) |
Рецептор |
Π |
JAK1.2; TYK2 |
1,3, 5a/b |
— |
||
тпо |
CD110 |
|
|
JAK2, TYK2 |
1,3, 5a/b |
— |
|
LIF, OSM |
LIF/OSMR, CD130(gpl30) |
JAK1, 2; TYK2 |
1,2,3 |
gpl30 |
|||
C N T F |
CNTFR2 , |
CD130(gpl30) |
JAK1, 2; TYK2 |
1,2,3 |
gpl30 |
||
и рецепторы подсемейства ИЛ-2 (см. табл. 3.7), часто передают сигнал про тивоположной полярности — ИЛ-10 ингибирует транскрипцию тех генов, которые другие цитокины активиру ют, действуя через тот же набор STAT-белков. Опять же это, на пер вый взгляд, парадоксальное явление имеет простую бытовую аналогию: ес ли схема электрической проводки та кова, что освещение может быть включено из двух мест, то одно и то же движение тумблера может приво дить в одних случаях к включению света, а в других — к выключению.
Следует подчеркнуть, что инфор мации, приведенной в табл. 3.7—3.9, недостаточно для полного объяснения специфичности действия цитокинов и их рецепторов на уровне активации транскрипции генов-мишеней.
3. Семейство рецепторов ФНО (в него входят и 6 рецепторов смерти, которые более подробно рассмотрены в разделе 2.5) и семейство рецепторов ИЛ-1Р/TOLL (см. табл. 3.8, 3.9). Оба семейства передают сигнал после рек рутирования в рецепторные комплек сы адаптерных молекул, которые не имеют киназной активности, но явля ются промежуточными звеньями для рекрутирования и активации других сигнальных молекул — киназ и каспаз. Особенностью обоих семейств яв ляется активация транскрипционных факторов семейства NF-κΒ и/или за пуск программированной клеточной смерти (эта проблема более подробно рассмотрена в разделе 3.4). Многие гены, активируемые при воспалитель ном ответе за счет действия ФНО, реагируют на активацию транскрип-
160
Т а б л и ц а |
3 8. Семейство рецепторов |
Ф Н О |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
Лиганд |
|
Другие назва |
|
Рецепторы |
|
Комментарии |
|||
|
ния |
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
TNF |
|
T N F а л ь ф а |
TNFRp55 |
(TNFRSF1A), |
|
He исключено, что p75 пере |
|||
|
|
|
TNFRp75 |
(TNFRSF1B) |
|
дает сигнал с помощью р55 |
|||
LTa |
|
T N F б е т а |
TNFRp55 |
(TNFRSF1A), |
|
|
— |
||
|
|
|
TNFRp75 |
(TNFRSF1B) |
|
|
|
|
|
LT бета |
|
|
LTбетаR (TNFRSF3) |
|
Активная форма — LTбета2LTальфа1 |
||||
OX40L |
|
|
OX40 |
(TNFRSF4) |
|
|
— |
||
CD40L |
|
Аро-1 |
CD40 (TNFRSF5) |
|
|
— |
|||
FasL |
|
|
Fas (CD95, TNFRSF6) |
|
Рецептор — "приманка" |
||||
|
|
|
DcR3 (TNFRSF6B) |
|
|
|
|
||
CD27L |
|
|
CD27 (TNFRSF7) |
|
|
— |
|||
CD30L |
|
|
CD30 (TNFRSF8) |
|
|
— |
|||
4-1BBL |
|
|
4-1BB |
(TNFRSF9) |
|
|
— |
||
TRAIL |
|
Apo-2L, TL2 |
DR4 |
(TNFRSF10A) |
|
Рецептор — "приманка" |
|||
|
|
|
DRS.fTNFRSFlOB) |
|
|
|
|
||
|
|
|
DcRl |
(TNFRSF10C) |
|
То же |
|
|
|
|
|
|
DcR2 (TNFRSF10D) |
|
|
|
|
||
TRANCE |
|
RANKL, |
RANK (TNFRSF11A) |
|
|
— |
|||
|
|
OPGL, O D F |
OPG (TNFRSF1 IB) |
|
|
|
|
||
TWEAK |
|
DR3LG, |
DR3 (TNFRSF12) |
|
Не исключено, что истинный |
||||
|
|
AP03L |
|
|
|
|
лиганд DR3 — не TWEAK |
||
APRIL |
|
|
BCMA |
|
|
|
|
|
|
|
|
|
TACI |
|
|
|
|
|
|
BAFF |
|
THANK, |
BAFF-R |
|
|
Основные |
физиологические |
||
|
|
BLYS, TALL1 |
BCMA |
|
|
функции |
BAFF осуществля |
||
|
|
|
TACI |
|
|
|
ются через BAFF-R |
||
LIGHT |
|
LTгамма, HVEM-L |
TNFRSF14 |
|
Основные |
физиологические |
|||
|
|
|
LTбетаR |
|
|
функции |
LIGHT осуществля |
||
|
|
|
|
|
|
|
ются не через LTбетаR |
||
Ectodysplasin |
|
|
Edar |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Т а б л и ц а 3.9. Семейство рецепторов И Л - I P / T O L L |
|
|
|
|
|||||
|
|
|
|
|
|
||||
Рецептор |
|
Лиганды или распознаваемые бактериальные компоненты |
|||||||
|
|
|
|
|
|
|
|
|
|
IL1R |
|
IL-1 |
|
|
|
|
|
|
|
IL18R |
|
IL-18 |
|
|
|
|
|
|
|
TLR1 |
|
Гетеродимеризуется с TLR4, лиганд неизвестен |
|
|
|||||
TLR2 |
|
Узнает липопротеины |
грамположительных |
бактерий, микобактерий |
|||||
|
|
и дрожжей, липотейхоевые кислоты, пептидогликан |
|
|
|||||
TLR3 |
|
Узнает двухцепочечную Р Н К |
|
|
|
|
|||
TLR4 |
|
Ассоциирован с |
CD 14, узнает липополисахариды и другие гликолипиды |
||||||
|
|
грамотрицательных бактерий |
|
|
|
|
|||
TLR5 |
|
Узнает белки бактериального жгутика |
|
|
|
|
|||
TLR6 |
|
Гетеродимеризуется с TLR4, узнает пептидогликаны |
|
|
|||||
TLR7 |
|
Природные лиганды неизвестны; активируется антивирусными препарата |
|||||||
|
|
ми — производными имидазохинолина |
|
|
|
|
|||
TLR8 |
|
Неизвестны |
|
|
|
|
|
|
|
TLR9 |
|
Неметилированные CpG последовательности бактериальной Д Н К |
|||||||
TLR10 |
|
Неизвестны |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
11 - 7908 Д. Г. Заридае |
16 1 |
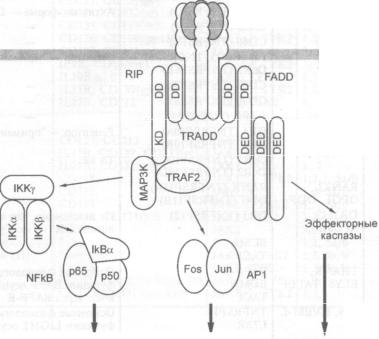
С х е м а 3.15. Передача активационного и апоптозного сигналов
через рецептор ФНО р55 (тример). Последовательность участия молекул RIP и TRAF2 в активации NFkB и API окончательно не установлена. МАРЗК не является киназой NIK, как это первона чально считалось. Не исключено, что активация каспазы 8 про исходит вне рецепторного комплекса
TNF
TNF-receptor
А к т и в а ц и я |
г е н о в |
Апоптоз |
ционного фактора NF - кВ . ЭТО не оз начает, что Ф Н О может активировать все гены, промоторы которых содер жат участок связывания NF - кВ; нуж но, чтобы все остальные условия, не обходимые для активации, были так же выполнены.
Наиболее полно изучена передача сигнала от рецептора Ф Н О р55 (TNFRp55) и FAS/Apo-1, относящих ся к категории рецепторов смерти.
Передача сигнала апоптоза более под робно рассмотрена в разделе 3.4, здесь же следует отметить важную анало гию, по-видимому, справедливую для многих рецепторов этого семейства. С помощью нескольких видов адаптер-
ных молекул (таких как TRAF, TRADD, FADD , RIP и др.) непосред ственно на агрегированных рецепто рах собирается многокомпонентный комплекс, существенный для переда чи сигнала (схема 3.15). В этот ком плекс, называемый иногда сигналосомой, входят как ферменты (протеинкиназы), так и субстраты соответст вующих реакций (дополнительные протеинкиназы, или транскрипцион ные факторы, или их ингибиторы, на пример ІкВ). В результате такой близ кой локализации субстратов последо вательных реакций достигается быст рая и эффективная передача сигнала, делающая эту группу рецепторов ни-
162
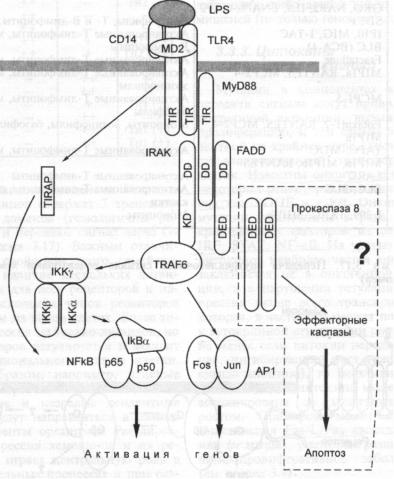
С х е м а 3.16. Передача активационного и апоптозного сигнала
через TOLL-рецептор (на примере TLR4). Точная стехиометрия этих рецепторов не известна. Возможность передачи апоптотического сигнала через прокаспазу 8 (область рисунка, обведенная
пунктиром) in vivo пока окончательно не установлена
чуть не менее эффективной, чем те рецепторы, у которых протеинкиназная активность содержится в поли пептидной цепи цитоплазматической части рецептора или в молекулах ас социированных с рецептором киназ типа JAK. Кроме того, подобная орга низация сигнальных молекул в еди ный комплекс повышает надежность и специфичность передачи сигнала.
Сходные, но несколько отличные в деталях механизмы использования
11*
адаптерных молекул характерны для рецепторов ИЛ-1 и ИЛ-18. Оказалось, что эти два рецептора гомологичны эволюционно очень древнему семей ству TOLL (схема 3.16).
4. Семейство рецепторов для хемотаксических цитокинов (хемокинов) — самое моногочисленное и очень древ нее (табл. 3.10—3.11). Следует при знать, что еще 10 лет назад физиоло гическая роль хемотаксиса лейкоци тов и других клеток иммунной систе-
163
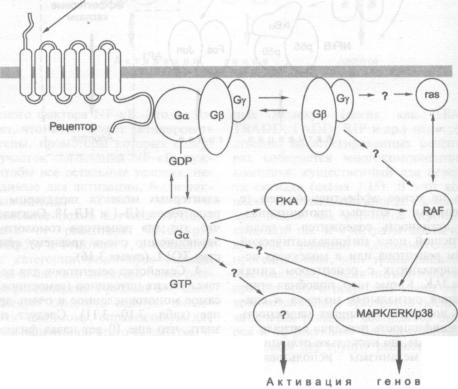
Т а б л и ц а |
3.10. Рецепторы хемокинов |
|
|
|
|
|
|
Рецеп |
|
Лиганды |
Клетки, на которых экспрессируется рецеп |
тор |
|
тор |
|
|
|
||
|
|
|
|
CXCR1 |
|
IL8 » G C P 2 » NAP2, ENA78 (в |
Нейтрофилы, нативные Т-лимфоциты |
|
|
порядке уменьшения сродства) |
|
CXCR2 |
|
G R O , NAP2, 1L8, ENA78, G C P 2 |
|
CXCR3 |
|
SDF1 |
Нейтрофилы, Т- и В-лимфоциты, моноциты |
CXCR4 |
|
IP10, M I G , I-TAC |
Активированные Т-лимфоциты, моноциты |
CXCR5 |
|
BLC (ВСА-1) |
B-лимфоциты |
CX3CR1 |
|
Fractalkine |
Активированные Т-лимфоциты, моноциты |
CCR1 |
|
M l P l a , RANTES, МСР2-4 |
Активированные Т-лимфоциты, моноциты, |
|
|
|
эозинофилы |
CCR2 |
|
МСР1-5 |
Активированные Т-лимфоциты, моноциты, |
|
|
|
базофилы |
CCR3 |
|
Eotaxinl-2, RANTES, MCP2-4, |
Моноциты, эозинофилы, базофилы |
|
|
M l P l a |
|
CCR4 |
|
TAPC, M D C |
Активированные Т-лимфоциты, моноциты |
CCR5 |
|
M l P l a , M l P l b , RANTES |
|
CCR6 |
|
LARC |
Активированные Т-лимфоциты |
CCR7 |
|
ELC, SLC |
Активированные Т-лимфоциты, дентритные |
|
|
|
клетки |
CCR8 |
|
1-309, TARC, M l P l b |
Моноциты |
|
|
|
|
С х е м а 3.17. Передача внутриклеточного сигнала рецепторами хемокинов
(мономер)
Хемокин
Т а б л и ц а |
3.11. Цитокины, |
продуцируе |
||||
мые Т-хелперами (мышь) |
|
|
|
|||
|
|
|
|
|
|
|
Цито- |
|
Продук |
Продук |
|
У чело |
|
ция: Тн1- |
ция: Тн2- |
|
||||
кмн |
|
века |
||||
|
клетки |
клетки |
|
|||
|
|
|
|
|
||
|
|
|
|
|
|
|
ИЛ-2 |
|
++ |
— |
|
Тн2 |
(+) |
ИФН-гамма |
|
++ |
— |
|
— |
|
ЛТ-альфа |
|
++ |
— |
|
— |
|
Ф Н О |
|
++ |
+ |
|
Тн2 |
( + + ) |
ГМ - КСФ |
|
++ |
+ |
|
Тн2 |
( + + ) |
ИЛ-3 |
|
++ |
++ |
|
— |
|
ИЛ-4 |
|
— |
++ |
|
— |
|
ИЛ-5 |
|
— |
++ |
|
— |
|
ИЛ-6 |
|
— |
+ |
|
Тн1 |
(+) |
ИЛ-9 |
|
— |
++ |
|
— |
|
ИЛ-10 |
|
— |
++ |
|
Т н 1 ( + ) |
|
ИЛ-13 |
|
— |
++ |
|
Т н 1 ( + ) |
|
|
|
|
|
|
|
|
мы была явно недооценена. Рецепто ры хемокинов содержат 7 трансмем бранных доменов (гомологичны ро допсину) и передают сигнал через G- белки (схема 3.17). Важным отличи тельным свойством этого семейства является наличие нескольких лиган дов почти для всех рецепторов и на личие нескольких типов рецепторов на каждом из видов клеток. Более то го, экспрессия не только лигандов, но и рецепторов регулируется и зависит от функционального статуса клетки. Таким образом, например, наивные или активированные Т-клетки, а так же зрелые и незрелые дендритные клетки будут направляться в разные компартменты организма. Регулируе мая экспрессия хемокинов и их ре цепторов играет центральную роль в воспалительных процессах и при раз витии иммунного ответа.
3.3.2. Некоторые важнейшие цитокины
Перечислим некоторые важнейшие цитокины. С точки зрения противо опухолевого иммунитета, наиболее важными цитокинами следует считать факторы роста Т-клеток, такие как ИЛ-2, иммунорегуляторные цитоки ны, ассоциированные с типом Т-хел- перного ответа (ИФН-у, ИЛ-4, ИЛ-12,
ЛТ, ФНО , ИЛ-10 - см. табл. 3.11), а также Fas-L, CD40 и гемопоэтические факторы роста. Поляризация Т-хелпе- ров скорее всего связана с активацией специфического набора транскрипци онных факторов, которые включают либо один, либо другой набор геновмишеней (не только генов цитокинов).
3.3.3.Цитокины
и опухолевые клетки
Мутации в компонентах каскада передачи сигнала могут приводить к постоянной активации, вызывающей пролиферацию, и тем самым эквива лентной по крайней мере одному эта пу злокачественной трансформации клеток. Известны онкгенные формы
.некоторых рецепторов ц и т о к и н о в , таких как P D G F и c-Kit. Онкогенные
мутации установлены в ряде транс крипционных факторов из семейств IRF, STAT. NF-кВ. На уровне самих цитокинов наиболее частая аномалия заключается не в онкогенной мута ции, а в нарушении регуляции экс прессии (чаще всего транскрипции), которая, в частности, может привести к аутокринной стимуляции роста. Тем не менее если цитокин передает сиг нал, ингибирующий рост (например, сигнал апоптоза), то неактивная мутантная форма цитокина может быть ассоциирована со злокачественным ростом. Примером может служить инактивация Fas-L/Fas каскада в gld или Ipr мышах, у которых развивается лимфопролиферативное заболевание (см. раздел 3.4).
Некоторые цитокины, продуцируе мые клетками иммунной системы (как гемопоэтическими, развивающи мися из костного мозга, так и клетка ми стромы), оказывают воздействие на опухолевые клетки, в том числе действуя как факторы роста. Злокачественные клетки и сами способны Производить цитокины, и экспрессировать соответствующие рецепторы, так что в некоторых случаях в резуль тате аберрантной экспрессии одного из этих двух компонентов могут воз-
165
никать аутокринные или паракринные механизмы стимуляции роста опу холей.
Кроме того, цитокины, продуци руемые опухолевыми клетками, спо собны блокировать иммунный ответ как на уровне лимфоцитов (например, через ТТФ-бета), так и на уровне антигенпрезентирующих клеток (напри мер, через ИЛ-10).
3.3.4.Цитокины
и противоопухолевый надзор
Идея о том, что иммунная система постоянно следит за клетками собст венного организма и уничтожает спонтанно возникающие опухолеродные клетки, была высказана много лет назад. Специфический компонент та кого надзора, очевидно, связан с Т-клетками, распознающими опухоле вые антигены, в тех случаях, когда эти антигены презентируются. Если опу холевые клетки теряют способность к презентации (за счет потери экспрес сии молекул М Н С или нарушений в механизмах процессинга антигенов — двух наиболее распространенных ме ханизмов ускользания от надзора), то остается шанс, что они будут распо знаны и уничтожены НК-клетками. Некоторые цитокины (такие как Ф Н О и ИФН-гамма) являлись естественными кандидатами на роль в неспецифиче ском компоненте противоопухолево го надзора, поэтому активное изуче ние цитокинов и развитие технологии генетического нокаута недавно приве ли к значительным успехам в разра ботке и проверке предположений этой теории.
Недавние эксперименты с нокаутными мышами указали на централь ную роль в иммунологическом надзо ре ИФН-гамма. Во-первых, мыши, в кото рых инактивация ИФН-гамма сочеталась с инактивацией р53, погибали от спон танных опухолей гораздо быстрее, чем р53-/- мыши. Во-вторых, у мышей с инактивацией разных компонентов цепи передачи сигнала (ИФН-гамма, его
рецептора или транскрипционного фактора STAT-1) индуцированные и спонтанные опухоли возникали гораз до раньше и чаще, чем у контрольных мышей. Такое же явление наблюда лось у мышей с инактивацией генов rag (у таких мышей невозможны пере стройка генов иммуноглобулинов и Т- клеточного рецептора, поэтому они не обладают функциональными лимфо цитами). Результаты исследований оз начают, что ИФН-гамма является цен тральным регулятором противоопухо левого надзора, а саму функцию над зора осуществляют лимфоциты. Дру гое важное наблюдение, сделанное в ходе этих исследований, состоит в том, что спонтанные опухоли, возни кающие в иммунодефицитных мышах, гораздо более иммуногенны, чем опу холи, возникшие у контрольных мы шей. При трансплантации опухолей иммунодефицитных мышей реципи ентам с функционирующей иммунной системой такие опухоли легко оттор гаются, в то время как значительная часть опухолей мышей дикого типа сохраняет способность к агрессивному росту. Эти данные объясняются явле нием иммунного надзора, при котором происходит отбор клонов опухолевых клеток, не замечаемых иммунной сис темой. Опухолевые клетки, развиваю щиеся у иммунодефицитных мышей, не испытывают этого селективного давления. На основании этих наблю дений недавно была сформулирована концепция иммуноредактирования по пуляции опухолевых клеток в процес се иммунного надзора.
3.3.5. Цитокины в противоопухолевой
терапии. Применение цитокинов in vitro
Получение рекомбинантных цито кинов не только привело к настояще му прорыву в исследованиях биологи ческой функции и механизмов пере дачи сигналов, но также сыграло важ нейшую роль в медицине, в том числе в онкологии. Применение иммуномо-
166
дуляторных цитокинов в клинике мо жет рассматриваться как один из ва риантов неспецифической иммуноте рапии. Доступность таких цитокинов, как ИЛ-2, позволила культивировать линии и клоны Т-клеток, в частности цитотоксических Т-клеток, узнающих опухолевые антигены. Другие цитоки ны, такие как ИЛ-4 и ГМ - КСФ, при меняют для получения дендритных клеток из клеток костного мозга или крови. И те, и другие виды клеток ис пользуют в нескольких протоколах адаптивной иммунотерапии, при ко торых клетки после манипуляций in vitro (для увеличения их числа, акти вации и внесения дополнительного генетического материала) переносят обратно в организм пациента. Кроме того, в протоколах терапии опухолей используют цитокины, способствую щие дифференцировке и активации дендритных клеток.
3.3.6. Противоопухолевое применение цитокинов in vivo
Результаты системного или мест ного применения цитокинов для лече ния онкологических заболеваний весьма неоднозначны. Эта область клинической науки стала свидетелем как достижений, так и горьких раз очарований.
Оказалось, что в большинстве слу чаев предклинические испытания ци токинов, проведенные на животных, не позволяют правильно предсказать клинические результаты. Те цитоки ны, которые имеют выраженное про тивоопухолевое действие на мышах (даже при однократном системном применении), не дают ожидаемых те рапевтических эффектов у пациентов (в нетоксических дозах). Основная проблема, по-видимому, кроется в плейотропности действия многих ци токинов, в связи с чем при их систем ном применении затрагиваются не только те компартменты, в которых повышенная концентрация данного цитокина может дать терапевтический
эффект, но и те, где эффект может быть негативным. Такие негативные эффекты усиливаются вследствие взаимосвязанности биосинтеза мно гих цитокинов (на уровне экспрессии генов самих цитокинов или их рецеп торов), что может приводить к неже лательным последствиям. На уровне организма негативные эффекты цито кинов проявляются в системной ток сичности, которая была отмечена для большинства цитокинов на первых же стадиях клинических испытаний, что во многих случаях не позволило дос тичь терапевтических доз. К глубоко му сожалению, в этот список попали
исамые многообещающие цитокины, такие как ИЛ-2, Ф Н О , ИФН-гамма, ИЛ-12
имногие другие. Для объективности следует сказать, что интерфероны ти па 1 (ИФН-альфа и -бета) нашли ограничен ное применение при лечении ряда опухолей, имеются также сообщения об отдельных успехах в лечении опу холевых заболеваний с использовани ем ИЛ-2, Ф Н О и некоторых других цитокинов.
Системное (внутривенное) приме нение цитокинов последней группы сопряжено со значительной токсично стью, широко не используется и вряд ли будет применяться в качестве единственного агента, однако некото рые варианты комбинированной тера пии с использованием цитокинов попрежнему проходят клинические ис пытания. Так, ИЛ-2 в малых дозах яв ляется компонентом некоторых схем иммунотерапии, в том числе с ис пользованием адаптивного переноса специфических Т-клеток. Комбина ция Ф Н О , ИФН-гамма и химиотерапии успешно применяется для лечения опухолей конечностей при введении препаратов методом перфузии.
Следует иметь в виду, что цитокинотерапия совершенно не обязательно направлена непосредственно на опу холевые клетки: во многих случаях противоопухолевый эффект связан с прямым или опосредованным дейст вием на сосуды, питающие опухоль.
Нетоксичными и относительно эф -
167
фективными оказались интерфероны типа 1 при лечении волосатоклеточного лейкоза, карциномы мочевого пузыря, саркомы Капоши и в мень шей степени рака почки и ряда других онкологических заболеваний. С точки зрения онкологических больных, по лучивших положительный эффект от цитокинотерапии, лучшими оказались препараты Г - КСФ и эритропоэтина. Эти два цитокина не оказывают пря мого противоопухолевого действия, и их эффект связан с восстановлением нормального уровня нейтрофилов и эритроцитов соответственно в ходе курсов лекарственной терапии. При менение этих цитокинов позволило повысить агрессивность и эффектив ность курсов химиотерапии и умень шить количество нежелательных по бочных эффектов, таких как анемия и нейтропения. Неожиданным побоч ным эффектом развития цитокиноте рапии явилось их применение в спор тивной медицине, где некоторые пре параты цитокинов (например, эритропоэтин) квалифицированы как до пинг.
Из цитокинов, имеющих прямой противоопухолевый эффект, перспек тивным остается и аналог Ф Н О TRAIL/AP0-2L (см. табл. 3.8 и раздел 3.4), поскольку сведения о его токсич ности в отношении нормальных гепатоцитов не подтвердились. В стадии клинических испытаний находится еще ряд комбинированных протоко лов с применением цитокинов. Следу ет еще раз подчеркнуть, что в отличие от иммунотерапии с использованием антител или цитотоксических Т-кле ток, специфичных для конкретных опухолевых антигенов, цитокины про являют относительно неспецифиче ские эффекты, поэтому их будущее ограниченное применение скорее все го будет происходить в комбинации с другими методами иммунотерапии или лекарственной терапии. Так, ИЛ- 2 в малых дозах применяют в комби нации с адоптивным переносом цитолитических Т-клеток.
К другим возможностям примене
ния цитокинов в онкологической кли нике относятся более сложные попыт ки использовать иммунизацию паци ентов собственными опухолевыми клетками, которые были трансфицированы конструкциями для экспрес сии некоторых цитокинов (ИЛ-2, Г М - К С Ф и некоторых других). В этом случае цитокины могут усиливать спе цифический иммунный ответ.
Рекомендуемая литература
Darnell J. Е. Jr., Kerr I. M., Stark G. R. JakSTAT pathways and transcriptional activa tion in response to IFNs and other extra cellular signaling proteins // Science. — 1994. - Vol. 264. - P. 1415-1421.
Medzhitov R., Janeway C. Innate immune rec ognition: mechanisms and pathways // Immunol. Rev. - 2000. - Vol. 173. - P. 8 9 - 9 7 .
O'Garra A., Aral N. The molecular basis of T helper 1 and T helper 2 cell differentia tion // Trends Cell. Biol. - 2000. - Vol. 10. - P. 542-550.
Rane S. G., Reddy E. P. Janus kinases: com ponents of multiple signaling pathways // Oncogene. - 2000. — Vol.19. - P. 5662-5679.
Shankaran |
V., |
Ikeda H., |
Bruce |
A. T. |
et |
al. |
||
IFN-gamma and lymphocytes prevent pri |
||||||||
mary tumour development and shape tu |
||||||||
mour |
immunogenicity |
// |
Nature. |
— |
||||
2001. - Vol. 410. - P. 1107-1111. |
|
|||||||
Wallach D., |
Varfolomeev |
E. E., |
Malinin |
N. L. |
||||
et al. Tumor necrosis factor |
receptor |
and |
||||||
Fas signaling mechanisms // Annu. Rev. |
||||||||
Immunol. |
- |
1999. - |
Vol. |
17. - P. |
331. |
|||
3.4. Механизмы активации программируемой клеточной гибели через рецепторы смерти
С. А. Недоспасов, И. Н. Лаврик
Способность запускать самоликви дацию, или апоптоз, является неотъ емлемым свойством клеток высших эукариот. При этом виде клеточной гибели клетка разрушается вследствие коллапса внутриклеточных структур, но без разрыва клеточной мембраны (поэтому она может быть утилизиро вана фагоцитами, и даже массовая ги-
168
бель клеток не приводит к каким-ли бо патологическим процессам). У кле ток млекопитающих существует не сколько программ, запускающих кле точную гибель: в ответ на поврежде ния Д Н К , в ответ на отсутствие фак торов роста, а также в ответ на актив ный сигнал смерти, передаваемый секретируемыми или мембраносвязанными цитокинами. Именно по следний вид клеточной гибели, назы ваемый инструктивным апоптозом и передаваемый так называемыми ре цепторами смерти, и будет рассмотрен в этом разделе.
3.4.1.Рецепторы смерти
Первый рецептор этого вида был охарактеризован в 1989 г. в двух неза висимых исследованиях, в ходе кото рых моноклональные антитела к неиз вестной мембраносвязанной молеку ле, названной Аро-1, или Fas, вызыва ли программируемую клеточную смерть. Поскольку Ф Н О (фактор нек роза опухолей) вызывал цитотоксический эффект in vitro, а его рецепторы еще не были известны, то первона чально предполагалось, что эффект обусловлен одним из рецепторов ФНО, однако последующая очистка и клонирование Аро-1/Fas выявили ре цептор с мол. массой около 45 кДа, который не связывал ни Ф Н О , ни
родственный ему лимфотоксин (ЛТ). Практически одновременно был кло нирован истинный (первый из двух) рецептор р55 Ф Н О (табл. 3.12), при чем последующий тщательный анализ позволил выявить гомологичные до мены в цитоплазматических участках р55 и Аро-1/ Fas, мутации в которых
устраняли |
способность обоих рецеп |
|
торов |
вызывать апоптоз в ответ на |
|
Ф Н О |
или |
антитела к Apo-l/Fas. Этот |
белковый домен, длиной около 80 аминокислотных остатков, был назван "доменом смерти". Дальнейшие иссле дования и компьютерный анализ уста новили (на основании гомологии ме жду внеклеточными доменами рецеп торов) существование семейства, включающего в себя около 20 генов, имеющих гомологии с р55 и apol/fas (по альтернативным номенклатурам — cdl20a/tnfrs2 и cd95/tnfrs6, см. табл. 3.14). Семейство, кодируемое этими генами, называют суперсемейством рецепторов Ф Н О , в то время как р55 и Apo-l/Fas/CD95, а также еще 4 мембранных белка составляют подсе мейство, которое характеризуется на личием во внутриклеточной части ре цепторов домена смерти. Эту подгруп пу рецепторов и называют рецептора ми смерти. В рецепторах смерти выде ляют 3 основных домена: внеклеточ ный с высокой степенью гликозилирования и с присутствием цистеин-
Т а б л и ц а 3.12. Некоторые характеристики рецепторов смерти
|
Полный |
Размер домена смер |
Цистеин- |
Тканеспецифичность |
|
|
Рецептор смерти |
размер |
ти (аа), гомология с |
богатые |
экспрессии, коммента |
||
|
(аа) |
TNF-R1, % |
участки |
рии |
|
|
|
|
|
|
|
|
|
TNF-R1 (CD120a) |
455 |
80 (100 %) |
4 |
Большинство |
тканей, |
|
|
|
|
|
регулируется ИФН-гамма |
||
FAS (АРО-1, CD95) |
335 |
68 (25 %) |
3 |
Большинство |
тканей |
|
DR3 (АРО-3, |
417 |
62 (47 %) |
4 |
Лимфоциты, |
лимфоид- |
|
TRAMP, LARD) |
|
|
|
ные органы |
|
|
DR4 (TRAIL-R1, |
468 |
70 (30 %) |
2 |
Большинство |
тканей |
|
АРО-2) |
|
|
|
|
|
|
DR5 (TRAIL-R2, |
411 |
66 (30 %) |
2 |
Большинство |
тканей, |
|
KILLER) |
|
|
|
регулируется р53 |
||
DR6 |
613 |
67(27 %) |
4 |
Большинство |
тканей |
|
|
|
|
|
|
|
|
169
богатых участков, трансмембранный и цитоплазматический. В табл. 3.12 приведены свойства 6 известных функциональных рецепторов смерти. Хотя в исходной номенклатуре для Fas/Apo-1 и TNFR1 использовались обозначения DR1 и DR2 соответст венно, эти термины практически не используют в научной литературе.
3.4.2. Цитокины, активирующие рецепторы смерти
Первыми идентифицированными цитокинами, способными вызывать клеточную гибель, опосредованную рецепторами смерти, оказались Ф Н О и ЛТ-альфа (следует отметить, что in vivo ЛТ-алфьа в комплексе с ЛТ-р действует через рецептор ЛТ-бета, не содержащий домена смерти, см. раздел 3.3). Основ ная форма Ф Н О — секретируемая, от части поэтому он гораздо лучше изу чен. Ф Н О — плейотропный цитокин со множеством специфических функ ций в иммунорегуляции и врожден ном иммунитете. Другая форма био логически активного Ф Н О — мембраносвязанная — находится на поверх ности клетки-продуцента. В этом слу чае для передачи сигнала через рецеп
тор нужен контакт с клеткой-мише нью. Аналогично суперсемейству ре цепторов Ф Н О , ЛТ и некоторые дру гие молекулы составляют суперсемей ство ФНО-подобных лигандов, при чем большую их часть представляют собой мембраносвязанные молекулы (см. раздел 3.3). Одной из молекул этого суперсемейства и оказался при родный лиганд (цитокин) для Аро-1/ Fas/CD95, который принято называть FasL (или Аро-1 L/CD95L). Биологи ческое значение этого мембраносвязанного цитокина было установлено с помощью мутантной линии мышей, отличающейся врожденным лимфопролиферативным нарушением (так называемые gld мыши). Аналогичная врожденная аномалия характеризует и Ipr мышей, у которых инактивирован Fas (см. 3.3.7).
Позднее были выявлены лиганды и для рецепторов DR4-DR5. В обоих случаях им оказался мембраносвязанный цитокин TRAIL/Apo-2L. Недавно был определен лиганд для D R 3 - U H T O - кин TL1A. Лиганд для DR6 пока не определен.
Кроме того, TRAIL/Apo-2L может связывать растворимые или мембра носвязанные рецепторы DcRl и D c R l , не способные передавать сиг-
Т а б л и ц а 3.13. Лиганды инструктивного апоптоза и их взаимодействие с рецепторами, в
том числе отличными от рецепторов смерти
Лиганд, альтер |
Рецептор, альтерна |
Передача |
Комментарий |
||
нативные на |
сигнала |
||||
тивные названия |
|||||
звания |
смерти |
|
|||
|
|
|
|||
|
|
|
|
|
|
Ф Н О (TNF) |
TNF-R1 |
(CD 120а) |
+ |
|
|
или лимфоток- |
TNF - R2 |
(CD120b) |
— |
При передаче сигнала TNF-R2, воз |
|
син-альфа (TNF-бета) |
|
|
|
можно, взаимодействует с TNF-R1 |
|
FASL (АРО-1 L, |
FAS (АРО-1, CD95) |
+ |
При передаче сигнала FAS, возмож |
||
CD95L) |
DcR3 (TR6) |
— |
но, взаимодействует с TNF-R1 |
||
TL1A |
DR3 (АРО-3, |
+ |
Имевшееся в литературе утвержде |
||
|
TRAMP, |
LARD) |
|
ние, что лиганд DR3 — TWEAK, по- |
|
|
DcR3 (TR6) |
— |
видимому, ошибочно |
||
TRAIL |
DR4 (АРО-2) |
+ |
|
||
(APO-2L) |
DR5 |
|
+ |
|
|
|
DcRl |
|
— |
|
|
|
DcR2 |
|
— |
|
|
Не определен |
DR6 |
|
+ |
— |
|
|
|
|
|
|
|
170
нал. Аналогичным образом FasL/Apo1L и TL1А могут взаимодействовать с DcR3 без передачи сигнала. Неактив ные аналоги рецепторов могут играть роль антагонистов сигнала клеточной гибели и защищать клетку от апоптоза. Соответствие природных цитокиновиндукторов клеточной гибели и ре цепторов смерти показано в табл. 3.13.
3.4.3. Каспазы
Каспазами называют особое семей ство субъединичных протеаз, которые характеризуются присутствием цистеинового остатка в активном центре, а также тем, что они расщепляют по липептидную цепь после остатка аспарагиновой кислоты. Каспазы синте зируются в виде неактивных предше ственников (зимогенов), их активация происходит после саморасщепления
предшественника, в результате чего образуются две субъединицы активно го фермента; полный фермент пред ставляет собой тетрамер из двух видов субъединиц (схема 3.18). Важно, что каскад расщеплений происходит толь ко после того, как молекулы предше ственника входят в состав многоком понентного белкового комплекса, ко торый и создает необходимые стерические условия для самоактивации.
При апоптозе активированные кас пазы протеолитически расщепляют ряд существенных структурных ком понентов клетки (называемых иногда субстратами апоптоза), инактивируют ряд важных белков и, наоборот, акти вируют дополнительные компоненты апоптозного механизма (например, специальную нуклеазу, которая при водит к быстрой деградации ядерной Д Н К ) . Общая схема инструктивного
С х е м а 3.18. Активация каспаз на примере каспазы 8. Расщеп
ление по остаткам Asp приводит к образованию субъединиц р20 и р10, формирующих активный фермент каспазы 8
DED-домен для рекрутирования
171
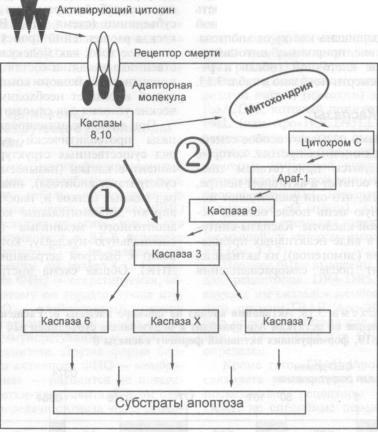
С х е м а 3.19. Два главных сигнальных каскада, индуцирующих
программированную клеточную гибель
Т а б л и ц а 3.14. Некоторые клеточные субстраты каспаз (субстраты апоптоза)
Название белка |
Сайт рас |
Кас |
Функциональная |
роль |
|
|
щепления |
паза |
|
||||
|
|
|
|
|
||
|
|
|
|
|
|
|
PARP |
D E V D * G |
3,7 |
Репарация Д Н К |
|
|
|
Ul-70kDa |
D G P D * G |
3,7 |
Сплайсинг Р Н К |
|
|
|
Gas2 |
SRVD*G |
? |
Компонент системы микротрубочек |
|||
ДНК - зависимая протеинкиназа |
D E V D * N |
3,7 |
Репарация Д Н К |
|
|
|
Протеинкиназа С сигма |
D M Q D * N |
3,7 |
Активируется при апоптозе, |
кон |
||
|
|
|
денсация |
ядра |
|
|
Фактор диссоциации D4-ГДФ |
D E L D * S |
3 |
Регуляция |
Rho-ГТФаз |
|
|
Ламин А |
V E I D * N |
6 |
Структурный белок ядерной |
обо |
||
|
|
|
лочки |
|
|
|
Фодрин |
D E T D * S |
? |
Белок цитоскелета, ассоциирован |
|||
|
|
|
ный с мембраной |
|
|
|
Предшественник Д Н К а з ы CAD |
? |
3,7 |
Активация |
механизма, |
фрагмента |
|
|
|
|
ция Д Н К |
|
|
|
С 1 , С2-рибонуклеопротеины |
? |
3,7 |
Процессинг пре - мРНК |
|
|
|
|
|
|
|
|
|
|
172
Т а б л и ц а 3.15. Каспазы
Группа |
Каспаза |
Функциональ |
Субстратная спе |
Домен для рек |
|
ная роль |
цифичность |
рутирования |
|||
|
|
||||
|
|
|
|
|
|
I |
1 |
Процессинг |
W E H D |
CARD |
|
|
4 |
цитокинов |
W E H D |
CARD |
|
|
5 |
|
W E H D |
|
|
|
13 |
|
W E H D |
|
|
II (каспазы-ис- |
2 |
Апоптоз |
D E H D |
CARD |
|
полнители |
3 |
|
DEVD |
|
|
апоптоза) |
6 |
|
VEHD |
|
|
|
7 |
|
DEVD |
|
|
|
CED - 3 (С. elegans) |
|
DEVD |
CARD |
|
III (регулятор- |
8 |
Апоптоз |
LETD |
D E D |
|
ные каспазы) |
ON |
|
L E H D |
CARD |
|
|
|
||||
|
10 |
|
LEXD |
D E D |
|
|
|
|
|
|
апоптоза показана в левой части схе мы 3.19. Важно, что эффекторная фа за этого механизма перекрывается или даже совпадает с таковой для второго механизма апоптоза, включающего в себя активацию митохондрий, высво бождение цитохрома С и активацию каспазы 9 (правая часть схемы 3.18). Каким образом осуществляется регу ляция этих двух основных путей апоп тоза, пока окончательно не установле но, но, как показано на схеме 3.19, даже на начальных стадиях эти меха низмы не полностью разобщены.
Некоторые субстраты каспаз при ведены в табл. 3.14.
Поскольку апоптоз — это эволюционно древний процесс, имеющий место уже у нематоды, то прародите лем каспаз высших организмов можно считать продукт гена ced3 из С. ele gans, однако у нематоды есть только один механизм апоптоза и отсутствует инструктивный апоптоз, инициируе мый рецепторами смерти. Неудиви тельно поэтому, что у человека и жи вотных охарактеризовано уже 14 кас паз, причем CED - 3 аналогична каспазе 3. Эта каспаза наряду с каспазой 7 непосредственно расщепляет субстра ты апоптоза, а ее активация является следствием активации дополнитель ных каспаз, таких как каспазы 8 и 9, иногда называемые регуляторными, или инициаторными, каспазами
(табл. 3.15). Выделяют три основные группы каспаз исходя из их субстрат ной специфичности. Интересно, что при этом каспазы с одинаковой спе цифичностью обладают сходной функциональной активностью. Ис ключение составляет каспаза 6, кото рая по структуре активного центра должна находиться в группе III, хотя по функции она относится к испол нителям апоптоза.
3.4.4. Адапторные молекулы рецепторов смерти и каскад сигналов, активирующих каспазы
Отличительной чертой механизма передачи сигнала рецепторами смерти является использование особого клас са адаптерных молекул — таких, как FADD, которые рекрутируются в ре цепторный комплекс за счет гомотипических белок-белковых взаимодей ствий. Считается, что для передачи сигнала рецепторы семейства Ф Н О должны образовывать димеры или тримеры (тримерная форма наиболее согласуется с тримерной конфигура цией лигандов, таких как Ф Н О , ЛТ и TRAIL/Apo-2L). Цитоплазматическая часть рецепторов смерти содержит до мены смерти — структурные блоки, склонные к гомотипической олигомеризации, поэтому адаптерные молеку-
173
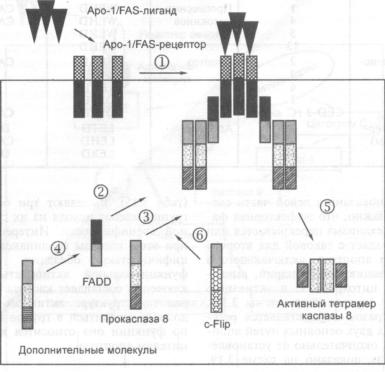
С х е м а 3.20. Образование белкового комплекса (DISC), запус
кающего апоптоз, на примере Apo-l/Fas/CD95 (на схеме показа ны только основные компоненты)
1 — связывание с Apo-l/Fas/CD95 лигандом и агрегация рецептора; 2 — рекрутирование FADD за счет взаимодействия "доменов смерти"; 3 — рекрутирование прокаспазы 8 за счет взаимодействия DED доменов; 4 — рекрутирование дополнительных молекул (прокаспазы 10. САРЗ), функ ция которых пока окончательно не установлена; 5 — саморасщепление и образование активного тетрамера каспазы 8; 6 — c-Flip может ингибировать процессинг прокаспазы 8 на уровне рекрутирования в инициаторный комплекс
лы, содержащие такой домен, и во |
мен этой молекулы — |
D E D — за счет |
|||||||||
влекаются в рецепторный комплекс. |
дополнительных |
|
гомотипических |
||||||||
Первичные |
адаптеры |
помогают рекру |
взаимодействий непосредственно рек |
||||||||
тировать |
в комплекс |
дополнительные |
рутирует |
в |
рецепторный |
комплекс |
|||||
белки, но за счет гомотипических |
предшественник |
одного |
из |
главных |
|||||||
взаимодействий между другими |
доме |
триггеров |
апоптоза |
— |
регуляторную |
||||||
нами, такими как DED - домены (см. |
прокаспазу 8. |
Самоактивация прокас |
|||||||||
табл. 3.14, 3.15 и схему 3.18). |
|
пазы 8 происходит без каких-либо до |
|||||||||
Наиболее |
полно |
изучен механизм |
полнительных ферментативных актив |
||||||||
передачи |
сигнала клеточной |
гибели |
ностей — лишь вследствие ее вовлече |
||||||||
через Apo-l/Fas/CD95 (схема 3.20). В |
ния в высокомолекулярный |
комплекс |
|||||||||
этом случае рецептор использует всего |
и создания оптимального стерическо- |
||||||||||
один тип адаптерных молекул — |
го контекста. В этом и состоит удиви |
||||||||||
FADD . |
Второй функциональный до- |
тельная простота |
этого механизма за- |
||||||||
174
пуска клеточной гибели. На следую |
3.4.5. |
Образование |
|
|
|||||||||||||||||||
щих |
этапах |
происходит |
активация |
|
многокомпонентного |
|
|
||||||||||||||||
каспаз 3 и 7, а затем и субстратов |
|
белкового |
комплекса |
(DISC), |
|||||||||||||||||||
апоптоза. |
|
|
|
|
|
|
|
|
|
|
|
запускающего |
механизм |
||||||||||
Аналогичный |
механизм |
действует |
|
клеточной |
гибели |
|
|
||||||||||||||||
при активации двух других рецепто |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
ров — DR4 и DR5 (под действием ли- |
|
Считается, |
что |
сборка |
активного |
||||||||||||||||||
ганда TRAIL/Apo-2L). В этих случаях |
инициаторного |
комплекса происходит |
|||||||||||||||||||||
также |
используется |
единственный |
вид |
за секунды после агрегации рецепто |
|||||||||||||||||||
адаптерного белка FADD, в результате |
ров под действием лигандов или анти- |
||||||||||||||||||||||
чего в комплекс вовлекается все та же |
тел-агонистов. Такой комплекс, назы |
||||||||||||||||||||||
прокаспаза 8. |
|
|
|
|
|
|
|
|
|
ваемый в литературе DISC, включает |
|||||||||||||
Сходный, |
но |
|
несколько |
более |
не |
только |
|
внутриклеточные |
домены |
||||||||||||||
сложный механизм установлен для ре |
смерти |
соответствующих |
рецепторов |
||||||||||||||||||||
цепторов |
TNFRp55 |
(активируемый |
и перечисленные |
ранее |
адаптерные |
||||||||||||||||||
ФНО и ЛТ-альфа) и DR3 (истинный ли |
молекулы и прокаспазу 8, но также |
||||||||||||||||||||||
ганд для этого рецептора окончатель |
целый |
ряд |
дополнительных |
факто |
|||||||||||||||||||
но не выявлен). В обоих случаях пер |
ров, |
функциональная |
роль |
которых |
|||||||||||||||||||
вичным адаптером, |
связывающимся с |
не полностью определена (табл. 3.16). |
|||||||||||||||||||||
доменами |
смерти |
во внутриклеточной |
Поскольку |
|
основные |
компоненты |
|||||||||||||||||
части |
рецепторов, |
|
является |
молекула |
комплекса не ассоциированы с ре |
||||||||||||||||||
TRADD, а вторичной адаптерной мо |
цептором до его взаимодействия с |
||||||||||||||||||||||
лекулой, которая вовлекается в ком |
лигандом, такая быстрая сборка и го |
||||||||||||||||||||||
плекс вслед за TRADD, является |
товность |
быстро |
включить |
сигнал |
|||||||||||||||||||
FADD (по-видимому, в олигомерном |
клеточной |
гибели |
предполагают осо |
||||||||||||||||||||
виде). Затем в этот комплекс привле |
бую |
компартментализацию |
компо |
||||||||||||||||||||
кается прокаспаза 8, хотя происходит |
нентов комплекса в цитоплазме. Не |
||||||||||||||||||||||
ли этот процесс на рецепторе, остает |
исключено, что частично "предсоб- |
||||||||||||||||||||||
ся пока неясным. |
|
|
|
|
|
|
|
|
ранный" комплекс |
адаптерных моле |
|||||||||||||
Такое |
распределение |
ролей |
между |
кул и прокаспазы 8 находится в неак |
|||||||||||||||||||
тивном |
состоянии |
и |
не |
может свя |
|||||||||||||||||||
адаптерными |
молекулами |
внутри |
все |
||||||||||||||||||||
заться с одиночными молекулами ре |
|||||||||||||||||||||||
го семейства |
рецепторов |
смерти |
по |
||||||||||||||||||||
цептора, однако агрегация |
рецепто |
||||||||||||||||||||||
зволяет определить |
белок FADD |
как |
|||||||||||||||||||||
ров |
создает |
дополнительные |
поверх |
||||||||||||||||||||
универсальный адаптер |
инструктив |
||||||||||||||||||||||
ности |
для |
|
гомотипических |
взаимо |
|||||||||||||||||||
ного апоптоза, а каспазу 8 — как уни |
|
||||||||||||||||||||||
действий, |
и такой |
комплекс рекрути |
|||||||||||||||||||||
версальный |
триггер |
инструктивного |
|||||||||||||||||||||
руется |
на |
агрегированный |
(тримери- |
||||||||||||||||||||
апоптоза. |
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Т а б л и ц а |
3.16. Состав многокомпонентного белкового комплекса, запускающего апоптоз |
||||||||||||||||||||||
через рецепторы смерти |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
Рецептор, на котором |
|
|
|
Адаптерные |
|
|
Прокас |
Дополнительные молекулы |
|||||||||||||||
собирается |
комплекс |
|
|
|
молекулы |
|
|
паза-8 |
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
TNFR1 |
(CD 120а) |
|
|
|
|
TRADD, |
FADD |
|
|
+ |
|
RIP, TRAF2 |
|
|
|||||||||
Fas (АРО-1, CD95) |
|
|
|
|
FADD |
|
|
|
|
+ |
|
Прокаспаза |
10, |
CAP 3. |
|||||||||
DR3 (АРО-3, TRAMP, |
|
|
TRADD, |
FADD |
|
|
+ |
|
RIP?, TRAF2? |
|
|
||||||||||||
LARD) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DR4(TRAIL-R1, АРО-2) |
|
|
FADD |
|
|
|
|
+ |
|
Прокаспаза |
10 RIP(?) |
||||||||||||
DR5 (TRAIL-R2, KILLER) |
FADD |
|
|
|
|
+ |
|
Прокаспаза 10 |
|
|
|||||||||||||
DR6 |
|
|
|
|
|
|
|
TRADD, |
FADD (?) |
|
(?) |
|
(?) |
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
175
зованный) рецептор. Структурные ас пекты этих взаимодействий сейчас проясняются с помощью рентгеноструктурного анализа.
3.4.6. Механизмы защиты клеток от инструктивного апоптоза
Исключительно быстрое образова ние инициаторного комплекса (DISC) и кажущаяся необратимость дальней шего каскада реакций, приводящих к расщеплению субстратов апоптоза, среди которых есть ключевые компо ненты, обеспечивающие целостность клеточных структур, ставят вопрос о механизмах контроля апоптоза. Выяс нилось, что несколько этапов в пере даче сигнала клеточной гибели могут быть заблокированы в результате ас социации специализированных белко вых ингибиторов с компонентами комплекса. В совокупности эти инги биторы предотвращают запуск апоп тоза вследствие случайной агрегации рецепторов и т. д.
Рассмотрим примеры регуляторных ингибирующих белков. Во-пер вых, в случае рецептора TNFR1 оха рактеризованы белки SODD, которые ассоциированы с внутриклеточными доменами смерти неагрегированных рецепторов и, по-видимому, стерически препятствуют их гомотипической агрегации. После агрегации внекле точных доменов рецепторов под дей ствием либо цитокина, либо агонистических антител белки SODD вы свобождаются из комплекса (вероят но, вследствие конформационных из менений в молекуле рецептора) и от крывают возможность для сборки инициаторного комплекса. Универ сальность этого механизма для других рецепторов смерти пока не продемон стрирована.
Кроме того, охарактризован белок c-FLIP, который рекрутируется в ре цепторный комплекс за счет наличия DED-домена, занимая позицию про каспазы 8. Тем самым ингибируются самоактивация прокаспазы и форми
рование активной каспазы 8, предот вращается запуск апоптоза.
Наконец, охарактеризовано семей ство c-IAP/XIAP-белков, которые свя зываются с каспазами и блокируют их процессинг/активацию. Эти белки способны блокировать как инструк тивный апоптоз, так и процессы са моликвидации клеток, запускаемые с участием митохондрий.
3.4.7. Роль инструктивного апоптоза in vivo
Роль описанных выше компонен тов механизма клеточной гибели в контексте целого организма была ус тановлена в трех типах эксперимен тов. Во-первых, были известны мутантные линии мышей с аномалиями, обусловленными генетическими де фектами в генах, кодирующих компо ненты инструктивного апоптоза, на пример Ipr и gld (табл. 3.17). Во-вто рых, аналогичные наблюдения были сделаны для пациентов с различными генетическими заболеваниями, на пример с врожденным лимфопролиферативным синдромом, связанным с делецией в гене fas/apol. И наконец, главную роль сыграли исследования с применением техники генетического нокаута для мышей; в этом случае удалось изучить и роль генов, мутации по которым детальны (см. табл. 3.17).
3.4.8. Роль инструктивного апоптоза при иммунном ответе на опухолевые клетки и в канцерогенезе
Fas экспрессируется на поверхно сти Т-клеток и играет активную роль в селекции Т-клеток в тимусе. Fas-L экспрессируется в иммунопривилегированных органах (например, в гла зах) и является частью механизма, устраняющего Т-клетки и делающего такие органы незаметными для им мунной системы. Вместе с тем Т-клет ки могут экспрессировать и лиганд, и рецептор апоптоза, причем считается, что Fas-L может отщепляться и дейст-
176
Т а б л и ц а 3.17. Физиологическая роль компонентов апоптозного каскада, определенная
на мутантных или нокаутных мышах
Белок, в гене ко |
|
Фенотип (в том числе свойства эм |
|
торого произошла |
Дефекты развития |
||
бриональных фибробластов, ЭФ) |
|||
мутация |
|
||
|
|
||
|
|
|
|
Fas-лиганд gld |
Отсутствие дефектов развития и |
Лимфопролиферативный син |
|
(мутация в С- |
роста |
дром, дефекты в селекции Т-кле |
|
концевом домене) |
|
ток в тимусе, аутоиммунные забо |
|
|
|
левания |
|
Fas-lpr (дефект |
Отсутствие дефектов роста и |
То же |
|
сплайсинга транс |
развития |
|
|
крипта гена Fas) |
|
|
|
TNFR1 |
Жизнеспособны без явных де |
Аномальная структура лимфоид |
|
|
фектов развития |
ных органов (селезенка, пейеровы |
|
|
|
бляшки), потеря врожденной за |
|
|
|
щиты от некоторых бактериальных |
|
|
|
и паразитарных инфекций |
|
DR3 |
Жизнеспособны без дефектов |
Дефекты селекции Т-клеток в ти |
|
|
развития |
мусе |
|
DR6 |
То же |
Гиперпролиферация Т-клеток в |
|
|
|
ответ на стимуляцию, повышенная |
|
|
|
секреция Тh2-цитокинов |
|
FADD |
Погибают через 11,5 дня эм |
Устойчивость ЭФ к апоптозу через |
|
|
брионального развития |
рецепторы смерти FAS, T N F R 1 , |
|
|
|
DR3 |
|
c-FLIP |
Погибают через 11,5 дня эм |
Повышенная чувствительность ЭФ |
|
|
брионального развития (дефек |
к апоптозу, индуцированному |
|
|
ты в развитии сердца) |
Ф Н О и Fas-лигандом |
|
Каспаза 8 |
То же |
Устойчивость ЭФ к апоптозу через |
|
|
|
рецепторы смерти |
|
Каспаза 9 |
Большая часть погибает из-за |
Нет активации каспазы 3 |
|
|
дефектов в развитии мозга, 2 % |
|
|
|
выживают |
|
|
Каспаза 3 |
Жизнеспособны (на C57BL6, но |
Не активируется каспаза 7, Т-клет- |
|
|
не выживают на 129-м бэкгра |
ки не погибают после активации |
|
|
унде). Спленомегалия |
анти-СDЗ |
|
|
|
|
вовать на соседние клетки. Тем не ме нее массового самоубийства Т-клеток не происходит, потому что этот про цесс активно блокируется внутрикле точными ингибиторами на одной из стадий передачи сигнала.
Интересно, что в некоторых случа ях опухолевые клетки экспрессируют Fas-L и потенциально могут элимини ровать цитолитические Т-клетки, во шедшие с ними в контакт. Общая зна чимость этого механизма, который может рассматриваться как один из примеров уклонения опухолевых кле ток от надзора со стороны иммунной
системы, пока однозначно не установ лена. Другим механизмом, используе мым опухолевыми клетками для бло кирования своей гибели, является экспрессия c-FLIP, причем в ходе развития опухоли в иммунокомпетентном хозяине происходит отбор опухолевых клеток с повышенным со держанием c-FLIP.
В последние годы разрабатывается такая важная проблема, как связь канцерогенеза с устойчивостью к апоптозу, приобретаемой опухолевы ми клетками на определенных стади ях. Так, мутации в генах fas и fast мо-
12-7908 Д. Г. Заридзе |
177 |
гут иметь прямое отношение в канце рогенезу, хотя общность этого явле ния надежно не установлена. Извест но, что потеря экспрессии Fas-L со провождает ранние стадии канцероге неза, вызванного ультрафиолетом. При раке предстательной железы в эпителиальных клетках детектируются мутации в гене fas, включающие инактивирующую мутацию в домене смерти. При некоторых других неоплазиях экспрессия Fas-L, наоборот, увеличивается, но одновременно клет ки приобретают устойчивость к апоп тозу через Fas за счет синтеза блокаторов апоптоза.
Известно, что одним из несколь ких механизмов цитотоксичности CD8+ Т-клеток по отношению к опу холевым клеткам или клеткам, зара женным вирусами, является передача сигнала от Т-клетки, экспрессирующей Fas-L, к клетке-мишени, экспрессирующей Fas. Экспрессия обоих видов молекул индуцибельна, и неко торые цитокины и терапевтические агенты способны ее модулировать.
3.4.9. Перспективы направленного использования инструктивного апоптоза в противоопухолевой терапии
Один |
из |
факторов инструктивного |
апоптоза |
— |
Ф Н О — был открыт как |
цитокин |
с |
противоопухолевым дейст |
вием и |
в |
течение долгого времени |
рассматривался как перспективный препарат для онкологической клини ки. Информация, приведенная здесь и в разделе 3.3, позволяет понять, поче му системное применение Ф Н О в ка честве единственного терапевтическо го агента не имеет перспективы. Вопервых, опухолевые клетки не имеют
избирательности |
на уровне экспрес |
сии рецепторов |
Ф Н О : первоначаль |
ные сообщения о том, что только ли нии опухолевых клеток чувствительны к Ф Н О , следует считать ошибочными. Во-вторых, рецептор T N F R 1 , экс-
прессируюшийся в больших количест вах практически на всех клетках, пе редает не только сигнал апоптоза, но и активационный сигнал, связанный с активацией транскрипционных фак торов и опосредованный молекулами RIP и TRAF2 (подробнее см. раздел 3.3). Этот активационный сигнал в за висимости от типа и статуса клетки может способствовать активации, пролиферации или дифференцировке, а также защите клетки от апоптоза. Таким образом, побочные эффекты от системного применения Ф Н О неиз бежны (в частности, эффекты, связан ные с провоспалительной активно стью ФНО), а результаты, полученные на нокаутных мышах, позволяют предположить, что в контексте целого организма Ф Н О скорее способствует, чем препятствует возникновению не которых видов опухолей. Казалось бы, Fas/Apo-1 и DR4-DR5 представляют собой более привлекательные мише ни, однако и здесь ситуация осложня ется отсутствием выраженной ткане вой специфичности этих рецепторов для опухолевых клеток. Более того, системное применение растворимого Fas-L, полученного с помощью техно логии рекомбинантной Д Н К , или агонистических антител может привести к гибели некоторых типов Т-клеток (как это было описано выше). Из ин дукторов апоптоза достаточно привле кательным остается рекомбинантный TRAIL/Apo2-L, поскольку его предклинические испытания (в том числе на обезьянах) не обнаружили систем
ной |
токсичности |
(следует |
отметить, |
что |
речь идет о |
препарате |
TRAIL/ |
Apo2-L, стабилизированном в форме тримера методами белковой инжене рии). Имеются указания на то, что опухолевые клетки экспрессируют го раздо больше рецепторов DR4-DR5, чем нормальные клетки. Вместе с тем окончательно не установлен относи тельный вклад прямого воздействия TRAIL/Apo2-L на опухолевые клетки и влияния на противоопухолевый эф фект других видов клеток (например, клеток эндотелия сосудов, которые
178
могут активироваться этим цитоки- |
этой |
теме |
растет |
экспоненциально, |
|||||||||||||||||||||||||||
ном). |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
достигнув |
уровня |
нескольких |
публи |
||||||||||||
Таким образом, |
механизмы |
избира |
каций в день. К 1995 г. выяснилось, |
||||||||||||||||||||||||||||
тельности |
действия |
|
лигандов |
инструк |
что |
активация |
теломеразы |
является |
|||||||||||||||||||||||
тивного апоптоза на опухолевые клет |
самым универсальным |
признаком |
ра |
||||||||||||||||||||||||||||
ки, которые могут определяться стату |
ковых клеток. Это значит, что теломе- |
||||||||||||||||||||||||||||||
сом других компонентов цепочки пере |
ры и теломераза могут стать основны |
||||||||||||||||||||||||||||||
дачи |
сигнала, |
представляют |
|
большой |
ми мишенями при терапии рака, а |
||||||||||||||||||||||||||
теоретический и практический интерес |
процесс канцерогенеза можно подраз |
||||||||||||||||||||||||||||||
и требуют дальнейшего |
изучения. |
|
|
делять на стадии в зависимости от те- |
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ломеразной активности. Кроме того, |
|||||||||||||||
Рекомендуемая литература |
|
|
|
|
|
|
теломеразная активность имеет значи |
||||||||||||||||||||||||
Krammer P. Н. CD95's |
deadly |
mission in the |
тельную |
диагностическую |
ценность |
и |
|||||||||||||||||||||||||
может |
использоваться |
|
для |
монито |
|||||||||||||||||||||||||||
immune |
system // |
Nature. |
— |
2000. |
— |
|
|||||||||||||||||||||||||
ринга процесса лечения. В то же |
вре |
||||||||||||||||||||||||||||||
Vol. |
407. |
- |
P. 789 - 795 . |
|
|
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
|
мя многообразие путей |
развития |
опу |
|||||||||||||||||||||||
Medema |
J. |
P., |
Scaffidi |
C, Kischkel |
F. |
C. |
et |
al. |
|||||||||||||||||||||||
холи |
приводит |
к |
усложнению |
общей |
|||||||||||||||||||||||||||
FL1CE is activated by association with the |
|||||||||||||||||||||||||||||||
картины, |
которая |
не |
может |
быть све |
|||||||||||||||||||||||||||
CD95 |
death-inducing |
signaling |
complex |
||||||||||||||||||||||||||||
дена |
к |
единой |
схеме. |
|
Сегодня |
идет |
|||||||||||||||||||||||||
(DISC) // Embo J. - |
1997. |
- |
Vol. |
16. |
- |
|
|||||||||||||||||||||||||
процесс |
накопления |
информации |
о |
||||||||||||||||||||||||||||
P. 2794-2804. |
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||
Nagata S. Apoptosis by death factor // Cell. — |
тканеспецифических |
различиях |
акти |
||||||||||||||||||||||||||||
1997. - Vol. 88. - P. 355-365. |
|
|
|
вации теломеразы в процессе канце |
|||||||||||||||||||||||||||
Nagata S. Human autoimmune lymphoprolif- |
рогенеза, идет поиск подходов к бло |
||||||||||||||||||||||||||||||
erative syndrome, a defect in the apopto- |
кированию |
теломеразы |
или |
использо |
|||||||||||||||||||||||||||
sis-inducing |
|
Fas receptor: |
a |
lesson |
from |
ванию |
теломеразы как |
мишени |
для |
||||||||||||||||||||||
the |
|
mouse |
model |
// J. Hum. Genet. — |
|||||||||||||||||||||||||||
|
борьбы |
с опухолями. Ниже |
будут рас |
||||||||||||||||||||||||||||
1998. - Vol. |
43. - |
P. 2 - 8 . |
|
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
смотрены |
особенности |
теломер |
и |
те |
|||||||||||||||||||||
Nagata |
|
S. |
Fas |
|
ligand-induced |
apoptosis |
// |
||||||||||||||||||||||||
|
|
ломеразы |
у |
человека, |
процесс |
канце |
|||||||||||||||||||||||||
Ann. Rev. Genet. — 1999. — Vol. 33. — |
|||||||||||||||||||||||||||||||
рогенеза с |
точки |
зрения |
|
реактивации |
|||||||||||||||||||||||||||
P. |
2 9 - 5 5 . |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
теломеразы, |
возможные |
подходы |
к |
|||||||||||||||||
Nagata S. Apoptotic DNA fragmentation // |
|||||||||||||||||||||||||||||||
Exp. Cell. Res. - 2000. - |
Vol. 256. |
- |
антителомеразной |
терапии рака. |
|
|
|||||||||||||||||||||||||
P. 12-18. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Nicholson D. W. Caspase structure, proteolytic |
3.5.1. История |
открытия |
|
|
|||||||||||||||||||||||||||
substrates, and function during apoptotic |
|
|
|||||||||||||||||||||||||||||
cell death // Cell. Death. Differ. - 1999. - |
теломеразы |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
Vol. 6. - P. 1028-1042. |
|
|
|
|
|
|
3.5.1.1. Смертные |
|
|
|
|
|
|
|
|||||||||||||||||
Zheng T. S., Hunot S., Kuida K., |
Flavell |
R. |
A. |
|
|
|
|
|
|
|
|||||||||||||||||||||
Caspase |
|
knockouts: |
matters |
of |
life |
and |
и бессмертные клетки |
|
|
|
|
||||||||||||||||||||
death // Cell. Death. Differ. — 1999. — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
Vol. |
6. |
- |
P. |
1043-1053. |
|
|
|
|
|
|
Более 100 лет назад, сразу после |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
возникновения |
методов |
|
длительного |
||||||||||||
3.5. Роль теломеразы |
|
|
|
|
культивирования |
клеток, |
|
появились |
|||||||||||||||||||||||
в |
канцерогенезе |
|
|
|
|
|
|
|
работы, |
авторы |
которых |
пытались от |
|||||||||||||||||||
|
|
|
|
|
|
|
ветить на вопрос о том, обладают ли |
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
Е. |
|
Е. |
Егоров |
|
|
|
|
|
|
|
|
|
клетки |
многоклеточного |
|
организма |
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
бесконечным пролиферативным |
по |
||||||||||||||
Всего 30 лет назад, в 1971 г., суще |
тенциалом. Среди множества работ |
||||||||||||||||||||||||||||||
ствование |
|
особого |
|
фермента, |
удли |
можно выделить две, убедительно по |
|||||||||||||||||||||||||
няющего |
концы линейных |
хромосом, |
казавшие, |
|
что |
соматические |
клетки |
||||||||||||||||||||||||
было |
|
предсказано |
|
А. М. |
Оловнико- |
могут |
быть |
практически |
|
бессмертны |
|||||||||||||||||||||
вым. В 1985 г. такой фермент, в даль |
ми. Каррель и его сотрудники культи |
||||||||||||||||||||||||||||||
нейшем получивший название тело- |
вировали фибробласты из сердца ку |
||||||||||||||||||||||||||||||
мераза, |
был |
экспериментально |
обна |
риного эмбриона в течение 34 лет. За |
|||||||||||||||||||||||||||
ружен. С тех пор количество работ по |
это время |
клетки |
прошли |
многие ты- |
|||||||||||||||||||||||||||
12* |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
179 |
||
сячи делений, не претерпевая сущест венных изменений в скорости роста и морфологии. Критики Карреля пред полагают, что благодаря несовершен ству методов культивирования экспе риментаторы могли постоянно вно сить свежие клетки в культуры и тем самым обеспечить постоянный рост. Другая, длительно проводившаяся ра бота начала XX в. лишена этого не достатка. Клетки асцитной опухоли культивировали в перитонеальных по лостях крыс в течение многих лет. Расчеты авторов показывают, что все клетки этой асцитной опухоли могли бы заполнить пространство от Солнца до Полярной звезды. Таким образом, с начала XX в. господствовало мне ние, что клетки многоклеточных орга низмов иммортальны, т. е. способны к бесконечно долгой пролиферации.
Наряду с такими успешными опы тами появлялось все больше и больше работ, выполненных в основном на клетках человека, авторы которых на блюдали плавное угасание пролифе рации клеток в культуре. Подобные результаты объясняли неадекватно стью методов культивирования и ошибками исследователей. В 1961 г. Хейфлик опубликовал результаты многолетних наблюдений, указывав шие на закономерности ограничения пролиферации в его опытах. Оказа лось, что культуры фибробластов че ловека способны расти более или ме нее определенное количество пересе вов (порядка 50). При этом культуры переживали определенные стадии рос та, морфология клеток закономерно менялась, и после остановки проли ферации клетки длительное время пе реживали в неделящемся состоянии.
После появления работ Хейфлика все клеточные культуры можно бы ло разделить на две группы: с огра ниченным пролиферативным потен циалом (смертные или мортальные) и иммортальные.
Оставался, однако, открытым во прос: насколько такое деление зави
сит от условий культивирования? Все гда можно было предположить, что какое-либо новое усовершенствова ние техники культивирования снимет ограничения пролиферативного по тенциала и клетки мортальные станут иммортальными. Для окончательного подтверждения этой классификации требовался фактический материал, объясняющий механизмы ограниче ния пролиферативного потенциала.
Помимо ограничения пролифера тивного потенциала, опыты Хейфли ка показали, что в ряде клеток суще ствует некий счетчик, ограничиваю щий общее количество делений, от пущенных данной клетке. После того как в общих чертах стали ясны меха низмы синтеза Д Н К , появилась ги потеза, предлагающая механизм ра боты такого счетчика. В 1971 г. А. М. Оловников предложил принцип маргинотомии в матричном синтезе полинуклеотидов, который заключается в том, что ДНК-полимераза не в со стоянии полностью реплицировать линейную матрицу, реплика получа ется всегда короче в начальной ее
части. |
Постепенное |
укорачивание |
Д Н К |
(недорепликация) |
ограничивает |
пролиферативный потенциал клеток и может служить основой "счетчика" клеточных делений в опытах Хейф лика.
3.5.1.2. Открытие теломеразы
Гипотеза Оловникова оставалась умозрительной, пока в 1985 г. Грейдер и Блекборн не обнаружили теломеразу. Выяснилось, что концы линейных хромосом простейшего Tetrahymena образованы короткой, тандемно по вторяющейся последовательностью Д Н К , которую синтезирует специаль ная полимераза (теломераза), имею щая постоянно ассоциированную с ней РНК-матрицу.
Теломеразы являются рибонуклеи новыми ферментами. РНК-компонент теломеразы содержит короткий район (матрицу), комплементарный одному повтору (часто матрица бывает длин-
180
нее, чем один повтор) G-богатой цепи теломерной ДНК .
Механизм действия теломераз — это повторное копирование матрицы, включающее этап элонгации, когда дезоксирибонуклеотиды последова тельно добавляются к З'-концу G-бо гатой цепи теломеры и транслокации фермента на конец новообразованной цепи. В результате действия теломера зы образуется достаточно длинный З'-конец, по которому затем достраи вается комплементарная цепь. В итоге теломера становится длиннее.
Было выяснено, что соматические клетки лишены теломеразной активно сти. Их теломеры укорачиваются как в процессе онтогенеза, так и при культи вировании клеток in vitro. В иммортальных, в том числе половых, клетках есть теломеразная активность и не происходит укорачивания теломер.
Теломеразы обнаружены в боль шинстве исследованных видов эука риот, имеющих линейные хромосомы. Оказалось, что теломеры подавляю щего большинства эукариотических хромосом образованы монотонно по вторяющимися короткими последова тельностями Д Н К , которые синтези руются при участии теломераз.
Теломераза способна не только поддерживать размер теломерного по втора, но и синтезировать теломеры de novo. Описано появление нормаль но функционирующих теломер de novo у человека. В ряде случаев а-талассе- мии происходит разрыв а-глобиново- го гена, расположенного на конце ма лого плеча 16-й хромосомы. При этом последовательность а-глобина непо средственно переходит в теломерную последовательность без образования каких-либо промежуточных субтеломерных последовательностей. Образо вание новых теломер таким способом означает, что наличие всего лишь од ного теломерного повтора является достаточным условием для образова ния функционирующей теломеры и что субтеломерные повторы у челове ка не несут теломероспецифических функций.
3.5.1.3. Теломерный повтор
Цитологи называют теломерами концевые участки хромосом, видимые в световом микроскопе. Это очень большие структуры, охватывающие районы в миллионы пар оснований Д Н К . Впервые описания теломер как особых структур, обеспечивающих це лость хромосом, появились в 1938 г. в работах Меллера и Мак Клинток. Суть этих ранних наблюдений заклю чается в том, что разорванная хромо сома остается нестабильной до тех пор, пока не обретет новую теломеру либо за счет рекомбинации, либо de novo.
Молекулярные биологи имеют де ло со значительно меньшим конце вым районом хромосомы, перекры вающим тысячи нуклеотидных пар. Именно с этими теломерами опериру ет теломерная теория и именно об этих теломерах идет речь в данной статье.
Подавляющее большинство изу ченных на сегодня хромосом имеют на конце многократно повторяющую ся монотонную последовательность. Это так называемый теломерный по втор. Субъединицей повтора обычно бывает последовательность длиной от 5 до 8 нуклеотидов. Непосредственно к теломерной последовательности (в хромосомах человека) примыкают по хожие на нее повторы, которые в сво ем большинстве имеют однонуклеотидные замены (по сравнению с теломерным повтором). В субтеломерной области имеются и повторы другого рода (по 61, 29, 37, 63, 75 и т. п.) нук леотидов. Субтеломерные повторы в отличие от теломерного обычно варь ируют от хромосомы к хромосоме и бывают хромосомоспецифичными.
Теломерный повтор — удивительно консервативная в эволюции струк тура.
Хромосомы всех позвоночных окан чиваются последовательностью TTAGGG, повторенной сотни и ты сячи раз.
181
Удивительно, но теломерная по следовательность T T A G G G встречает ся также у некоторых представителей членистоногих, червей и даже трипаносом. Очень похожие теломерные последовательности имеются у боль шинства растений — TTTAGGG, у ря да насекомых — TTAGG и нематод — TTAGGC .
Особые теломерные повторы, со стоящие из очень крупных субъеди ниц (сотни и тысячи нуклеотидов), имеются у хирономид (Chironomus), у некоторых лилейных (лук) и у ряда двукрылых (дрозофила). Считают, что такие теломерные повторы образуют ся без участия теломеразы.
У многих видов теломерные после довательности встречаются не только на концах хромосом, но могут быть расположены и во внутренних облас тях хромосомы. Например, у китай ского хомячка в геноме имеется 18 районов теломерных повторов внутри хромосом. Они расположены перицентромерно, по длине значительно превосходя теломерные повторы на концах хромосом. Хотя обычно внут ренние теломерные повторы локали зованы в перицентромерных областях, в некоторых случаях они локализуют ся в областях ядрышкового организа тора или около него. Происхождение и функции внутренних повторов не ясны. Наиболее распространено мне ние, что их появление связано с хро мосомными перестройками и поэтому отражает эволюцию кариотипа.
3.5.1.4. Тонкая структура теломер
Особый интерес в структуре теломер представляет их самая дистальная часть. Двойная спираль Д Н К не дохо дит до самого конца, 5'-цепь обрыва ется и остается одноцепочечный G-бо- гатый участок. Это свойственно всем теломерам, содержащим теломерные повторы, начиная от простейших и до человека. Длина одноцепочечного З'-конца варьирует от 10—16 нуклео тидов у простейших до нескольких со
тен нуклеотидов у человека. Стоит от метить, что свободный З'-конец име ется как в клетках с функционирую щей теломеразой, так и без нее, по этому его наличие невозможно объяс нить деятельностью теломеразы. В ли тературе не закончен спор о том, име ется ли одноцепочечный З'-конец на каждой теломере или же только у по ловины теломер. Если следовать тео рии недорепликации, то свободный З'-конец должен образовываться в ка ждом клеточном цикле только на од ной из двух теломер.
Наличие одноцепочечного фраг мента на каждой теломере означает, что его формирование идет путем де градации 5'-конца. Совсем недавно показано: длина одноцепочечного участка коррелирует со скоростью не дорепликации, что свидетельствует в пользу того, что деградация 5'-конца не вносит заметного вклада в укора чивание теломер. И наконец, показа но, что длина З'-конца может менять ся в процессе развития у растений: его длина различна в разных тканях.
Конформация одноцепочечного З'-конца привлекает внимание иссле дователей исходя из термодинамиче ских особенностей последовательно сти, содержащей повторяющиеся кла стеры G G G . Расчеты показывают, что такая последовательность может легко образовывать неканонические струк туры (триплексы, квадруплексы). Ре альным подтверждением возможности существования такой структуры яви лись опыты de Lange и соавт. На ос новании анализа вероятности появле ния ковалентных сшивок между нитя ми Д Н К была создана модель теломерных петель (схема 3.21). По этой модели свободный З'-конец в ком плексе с белками вступает во взаимо действие с двойной спиралью теломерной Д Н К и вытесняет одну из це пей (образуется тройной комплекс це пей Д Н К ) . Вытесненная цепь образует D-loop [вытесненную петлю (D — dis place)], а сама теломера при этом об разует теломерную петлю (t-loop). По данным Griffith и соавт., длина тело-
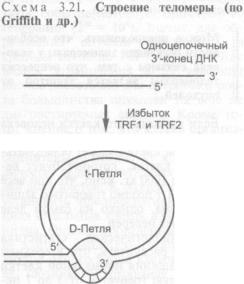
мерной петли коррелирует с длиной |
3.5.1.5. Теломеризация |
клеток |
|||||||||||||||
теломер, |
измеренной |
независимыми |
В |
конце 1997 г. удалось |
реконст |
||||||||||||
методами. |
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
руировать теломеразную активность в |
|||||||||
Одноцепочечная |
G-богатая |
тело- |
|||||||||||||||
лизате клеток. Оказалось, что для это |
|||||||||||||||||
мерная |
последовательность |
способна |
|||||||||||||||
го достаточно |
присутствия двух |
ком |
|||||||||||||||
образовывать |
внутрицепочечные |
и |
|||||||||||||||
понентов: каталитического компонен |
|||||||||||||||||
межмолекулярные |
квадруплексы. |
До |
|||||||||||||||
та теломеразы (TERT) и РНК-компо |
|||||||||||||||||
сих пор |
нет |
прямых свидетельств |
су |
||||||||||||||
нента (TERC). Следующим шагом бы |
|||||||||||||||||
ществования |
квадруплексов |
in |
|
vivo, |
|||||||||||||
|
ло введение TERT |
в клетки |
человека. |
||||||||||||||
однако |
разнообразные |
соединения, |
|||||||||||||||
Уже давно известно, что РНК-компо |
|||||||||||||||||
взаимодействующие |
с квадруплексами |
||||||||||||||||
нент |
экспрессируется |
в большинстве |
|||||||||||||||
in vitro, |
подавляют теломеразную ак |
||||||||||||||||
клеток человека независимо |
от |
тело- |
|||||||||||||||
тивность, |
в |
том |
числе |
и в |
клетках. |
||||||||||||
меразной активности, |
поэтому |
после |
|||||||||||||||
Найдено множество белков, специфи |
|||||||||||||||||
трансфекции |
клеток |
человека |
геном |
||||||||||||||
чески взаимодействующих с |
G- |
и |
С- |
||||||||||||||
T E R T |
в них |
появилась теломеразная |
|||||||||||||||
богатыми |
цепями |
теломерной |
Д Н К . |
||||||||||||||
активность. |
|
|
|
|
|
||||||||||||
Обнаружен фермент гираза, специфи |
|
|
|
|
|
||||||||||||
Введенная |
теломераза |
обладает |
|||||||||||||||
чески |
разрушающий |
квадруплексы. |
|||||||||||||||
Мутации этой гиразы вызывают син |
способностью |
поддерживать длину те |
|||||||||||||||
дром Блюма, |
характеризующийся |
це |
ломер — пролиферация клеток пере |
||||||||||||||
лым спектром изменений, начиная от |
стает угасать со временем и, по всей |
||||||||||||||||
специфических |
хромосомных |
пере |
видимости, клетки становятся иммор- |
||||||||||||||
строек и кончая внешним видом |
тальными. |
|
|
|
|
|
|||||||||||
больных. Существование таких белков |
В |
результате теломеризации клетки |
|||||||||||||||
доказывает, |
что ДНК в теломерах мо |
||||||||||||||||
жет иметь самую необычную конфор- |
не |
приобретают |
никаких признаков |
||||||||||||||
мацию, и эта конформация сущест |
злокачественной |
трансформации, а |
|||||||||||||||
венным образом влияет на функцио |
кариотип клеток |
остается |
стабиль |
||||||||||||||
нирование всей клетки. |
|
|
|
|
ным, как в культурах диплоидных |
||||||||||||
|
|
|
|
|
|
|
|
|
|
фибробластов человека. |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
Таким образом, |
теломерная теория |
||||||
|
|
|
|
|
|
|
|
|
|
старения клеток получила всесторон |
|||||||
|
|
|
|
|
|
|
|
|
|
нее признание. |
|
|
|
|
|||
3.5.1.6. Границы применимости теории недорепликации
|
В 1992 г. была предложена гипоте |
|
за недорепарации теломер как воз |
|
можного источника их укорачивания. |
|
Суть гипотезы в том, что поврежде |
|
ния, возникающие близко к концу |
|
хромосомы, хуже репарируются. Ги |
Связывание |
потеза может объяснять укорачивание |
различных |
теломер в неделящихся клетках, а так |
белков |
же в клетках без теломеразной актив |
|
|
|
ности. Гипотеза получила экспери |
|
ментальные подтверждения о преиму |
|
щественном накоплении повреждений |
Связывание |
в теломерах. |
TRF2 |
Известно, что низкое содержание |
183
кислорода увеличивает пролиферативный потенциал клеток, а при гипероксии резко снижается пролиферативный потенциал диплоидных фиб робластов человека.
Показано, что в условиях гипероксии диплоидные фибробласты челове ка теряют около 500 п.н. теломерного повтора за деление, что не может быть объяснено недорепликацией. Возмож но, большие потери теломерной Д Н К связаны с недорепарацией однонитевых разрывов, которые образуются и в нормальных условиях, но при гипероксии их количество в теломерах возрастает.
Таким образом, можно утверждать, что определенная часть случаев проли-
феративного старения клеток в куль туре не связана с недорепликацией те ломер. Вероятно, эти случаи во мно гом обусловлены несовершенством процессов культивирования.
3.5.2. Теломераза и опухоли
3.5.2.1. Роль репрессии теломеразы в соматических клетках
Можно считать установленным, что репрессия теломеразы ответствен на за старение клеток человека в куль туре (предел Хейфлика). В процессе пролиферации в опытах Хейфлика клетки значительно изменяются как морфологически, так и биохимически. Получены данные, свидетельствую щие о том, что похожие изменения клеток происходят in vivo, поэтому старение клеток в культуре можно рассматривать как процесс дифферен цировки. В этом случае репрессия те ломеразы — это механизм развития, при котором информация о пройден ных клеточных циклах записывается на Д Н К . Фактических данных в поль зу этой гипотезы пока очень мало, из вестно лишь, что терминальная дифференцировка и пролиферация прак тически всегда исключают друг друга, теломеры практически всех клеток ор ганизма короче, чем в клетках заро
дышевой линии, теломеразная актив ность иммортальных клеток быстро исчезает при искусственной дифференцировке в культуре, и при старе нии клеток в культуре в них появля ются маркеры дифференцировки.
Обращает на себя внимание тот факт, что теломераза человека, способ ее регуляции, надежность репрессии, процессивность фермента в бескле точной системе резко отличаются от других изученных теломераз (главным образом можно говорить о теломеразах мыши и других грызунов).
Спонтанная активация теломера зы в нормальных фибробластах чело века — событие необычайно редкое, в культуре фибробластов грызунов — явление рядовое, хорошо воспроизво димое.
Регуляция теломеразы человека, видимо, определяется ее белковой ча стью, но не РНК-компонентом; у мы шей скорее всего имеет место обрат ная закономерность.
Поскольку примерно 85 % опухо лей человека обладают теломеразной активностью, можно утверждать, что реактивация теломеразы участвует в онкогенезе. Следовательно, увеличе ние надежности репрессии теломера зы должно резко уменьшать вероят ность развития опухоли.
Можно предположить, что особен ности регуляции теломеразы у чело века связаны с тем, что репрессия теломеразы является защитой от опухолей.
Если принять, что клетки человека способны в среднем на 50 удвоений, то из одной клетки может образовать ся 2 • 1050 клеток, которые будут ве сить около 1000 кг. Ясно, что при весе опухоли в 1 т поздно говорить о защи те организма, однако на самом деле такой расчет неверен.
Эпидемиологическая статистика позволила рассчитать, что в среднем для превращения нормальной клетки в опухолевую требуется от 3 до 7 не зависимых случайных событий. Зна-
184
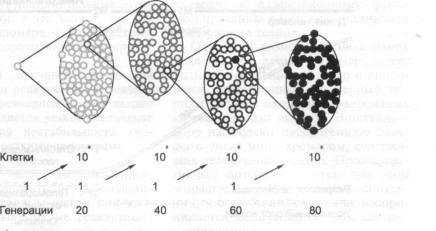
С х е м а 3.22. Последовательное клонирование при развитии опу
холи (по Harley и др.)
чит, при развитии опухоли происхо дит несколько этапов клонирования (схема 3.22). Процесс развития опухо ли в среднем включает от 3 до 7 мута ций. Если принять, что на одну мута цию приходится 20 генераций, то для образования опухоли требуется около 80 генераций.
Если считать, что в среднем часто та мутаций 1 на 106 клеток, то каждое такое событие потребует примерно 20 генераций (220 ~ 106). Значит, для об разования опухоли потребуется в среднем от 60 до 140 генераций. Как видно, ограничения в 50 удвоений вполне достаточно для остановки рос та большинства опухолей на еще не диагностируемых стадиях. Кроме то го, мнение о том, что клетка организ ма способна в среднем на 50 делений, сомнительно, поскольку к такому ко личеству делений способна только ма лая доля клеток. К сожалению, трудно установить пролиферативный потен циал отдельных клеток в составе орга низма. Опыты, проведенные в культу ре клеток, свидетельствуют, что даже внутри одного клона клеток существу ет (возникает) огромная гетероген ность клеток по пролиферативному потенциалу.
3.5.2.2. Процесс иммортализации клеток человека
Для объяснения процессов иммортализации клеток человека была пред ложена М1-М2 гипотеза (схема 3.23). Согласно этой гипотезе, для иммор-
тализащи клеткам необходимо пре одолеть два различных механизма, ог раничивающих пролиферацию. Первый механизм (M1) включается, когда длина теломер достигает некоего ми нимума. Именно Ml ответствен за старение клеток в культуре (предел Хейфлика).
До сих пор неясно, каким образом запускается M l , поскольку длина те ломер при этом составляет несколько тысяч нуклеотидов. По одной гипоте зе, относительно короткие теломеры способны служить в качестве сигнала повреждения Д Н К , по другой — уко рачивание теломер изменяет экспрес сию регуляторных генов, локализо ванных в субтеломерном гетерохроматине.
Белки различных опухолевых ДНК-содержащих вирусов способны увеличивать пролиферативный потен циал клеток человека и тем самым об-
185
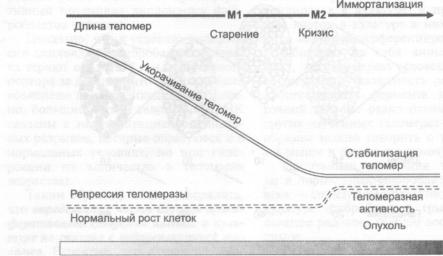
С х е м а 3.23. Процесс иммортализации фибробластов человека
(по Wright, Shay)
ходить (отменять) M l . Этими свойст вами обладают Т-антиген SV40, анти гены Е6/Е7 вируса папилломы чело
века типа 16, |
антигены E l a / E l b аде |
новируса типа 5. |
|
Различными |
способами показано, |
что в реализации Ml участвуют белки р53 и pRb или pRb-подобная актив ность. В эпителиальных клетках чело века Ml менее надежен, чем в фиб робластах; в его реализации не участ вует pRb. Пролиферативный блок при Ml опосредуется факторами типа р21, которые подавляют активность ком плексов циклин/cdK.
Обход (отмена) М1 позволяет клет кам пролиферировать дополнительное время (для фибробластов человека — около 30—40 удвоений популяции), пока второй механизм (М2) не вызо вет состояние кризиса. Кризис харак теризуется процессом пролиферации, сопровождающимся массовой гибе лью клеток. При этом размер популя ции какое-то время может быть ста бильным. С низкой вероятностью по сле кризиса могут возникнуть иммортальные клетки. Полагают, что кризис вызывается чрезмерным укорачивани
ем теломер, приводящим к их физиче скому исчезновению. В большинстве случаев единственным путем выхода из кризиса является реактивация те ломеразы.
3.5.2.3. Каким образом опухолевые клетки преодолевают репрессию теломеразы?
Частота спонтанной реактивации теломеразы в трансформированных SV40 диплоидных фибробластах чело века после прохождения ими допол нительных раундов репликации со ставляет 3 • 10-7—5 • 10-8.
В нормальных нетрансформированных фибробластах человека спон танная реактивация теломеразы (иммортализация) настолько редка, что ее невозможно воспроизвести в эксперименте.
Столь низкая вероятность указыва ет на то, что скорее всего реактивация теломеразы требует как минимум двух мутационных событий.
Все воспроизводимые случаи иммортализации фибробластов человека в культуре происходят после того, как клетки пройдут дополнительные раун ды репликации, а это значит, что ре активации теломеразы предшествует чрезмерное укорачивание теломер.
Возможной причиной увеличения вероятности реактивации теломера зы при чрезмерном укорачивании теломер является резкое увеличение генетической нестабильности хро мосом с короткими теломерами.
Учитывая все вышесказанное, можно предположить, что в большин стве случаев первым шагом при кан церогенезе являются не реактивация теломеразы, но другие события, по зволяющие клетке прежде всего полу чить селективное преимущество в росте и отменить механизмы старе ния. В пользу этой возможности сви детельствует также тот факт, что теломеры опухолевых клеток с реактиви рованной теломеразой почти всегда короче, чем теломеры из окружающей опухоль нормальной ткани.
3.5.2.4. Нетеломеразное удлинение теломер у человека
Около 5 % опухолей человека име ют независимый от теломеразы меха низм поддержания длины теломер. Этот механизм называют альтернатив ным (ALT). Полагают, что основу ALT составляют гомологичная реком бинация и генная конверсия. В клет ках, обладающих ALT, часто обнару живают специализированные образо вания, которые называют PML-тель- цами. В состав этих телец входят кус ки теломерной Д Н К , специфически связывающиеся с теломерным повто ром белки (обычно TRF1 и TRF2) и белки, связанные с рекомбинацией и репликацией.
Клетки, имеющие ALT, характери зуются очень гетерогенными теломе рами. Длина теломер может превы шать 50 т. п.н. При этом в клетке мо
гут находиться и относительно корот кие теломеры. Искусственное введе ние гена теломеразы в клетки с ALT приводит к одновременному функ ционированию в них двух механизмов поддержания теломер.
Одним из возможных механизмов поддержания длины теломер может быть рекомбинация теломер с эписомами, содержащими теломерный по втор. В случае заражения лимфоидных клеток человека вирусом Эпштейна— Барр наблюдают персистенцию боль шого числа мини-хромосом, содержа щих теломерный повтор. Пролифера тивный потенциал клеток при этом возрастает до 150 удвоений популя ции, что в обычных условиях воспри нимается экспериментатором как иммортализация.
3.5.3. Теломераза, теломеры и лечение опухолей
3.5.3.1.Значение теломеразы
вдиагностике опухолей
Прежде |
всего следует |
отметить, |
что наличие |
теломеразной |
активно |
сти может являться важным диагно стическим признаком. Приведем пример исследования теломеразной активности в нейробластомах. Из 100 исследованных опухолей 6 не содер жали теломеразной активности, 73 обладали низкой активностью, 21 — высокой. Большинство нейробластом с высокой теломеразной активностью имели дополнительные генетические изменения (амплификация N-тус и др.), которые отсутствовали в слу чаях со средней активностью. Ко
времени |
публикации |
погибло 12 |
больных, |
опухоли которых обладали |
|
высокой |
теломеразной |
активностью, |
2 больных со средней и ни одного — с опухолями без теломеразной актив ности. У 3 из 6 больных последней группы отмечена спонтанная регрес сия опухоли.
Доля опухолей, обладающих тело меразной активностью, зависит от ти па клеток. Например, теломеразной
187
активностью обладают около 10 % |
3.5.3.2. Подавление |
|
|
|||||||||
анапластических |
астроцитом, |
75 % |
теломеразы |
|
|
|
||||||
глиобластом и 100 % олигодендрогли- |
как метод лечения |
|
|
|||||||||
ом, 93 % форм рака молочной желе |
опухолей |
|
|
|
||||||||
зы, 80 % — рака легких. При мелко |
|
|
|
|
|
|
||||||
клеточном раке легкого почти в 100 % |
Как мы уже указывали выше, без |
|||||||||||
случаев обнаруживается высокая тело- |
активации |
теломеразы |
большинство |
|||||||||
меразная |
активность. |
|
|
|
опухолей не могло бы достигать ста |
|||||||
Корреляция |
теломеразной |
актив |
дий, опасных для здоровья. Можно |
|||||||||
ности |
со |
злокачественностью |
показа |
поэтому полагать, что подавление те |
||||||||
на также при раке желудка и толстой |
ломеразной |
активности, |
начатое во |
|||||||||
кишки, раке печени, опухолях пред |
время, могло бы помочь большинству |
|||||||||||
стательной железы, почек и кожи. |
онкологических |
больных. |
|
|
||||||||
Слабая |
теломеразная |
активность об |
Рассмотрим |
возможные |
последст |
|||||||
наружена при хроническом гепатите и |
вия терапии рака ингибиторами те |
|||||||||||
циррозе печени. |
|
|
|
|
ломеразы. Препараты, помимо опу |
|||||||
Огромные усилия, |
потраченные на |
холи, должны влиять на половые и |
||||||||||
изучение |
теломеразной |
активности |
стволовые клетки. У женщин такой |
|||||||||
как раннего маркера опухолей, свиде |
препарат не может повредить поло |
|||||||||||
тельствуют, что |
теломеразная |
актив |
вым клеткам, поскольку их проли |
|||||||||
ность не может являться самодоста |
ферация |
заканчивается |
в эмбриоге |
|||||||||
точным |
признаком |
для |
диагностики |
незе. У мужчин препарат должен |
||||||||
ранних |
стадий канцерогенеза. |
|
влиять |
на |
сперматогенез, |
однако |
||||||
Взависимости от происхождения криоконсервация спермы поможет
опухоли теломеразная активность имеет различное диагностическое зна чение. Если для одних типов опухолей уровень теломеразной активности яв ляется показателем длительной эво люции опухолевых клеток и, следова тельно, связан с плохим прогнозом, для других опухолей это не так. В конце XX в. имелось порядка 1000 публикаций, связывающих теломераз ную активность и свойства опухолей.
При диагностике опухолей следует учитывать и тот факт, что теломераз ная активность тесно связана с уров нем пролиферативной активности клеток. При переходе в пролифера тивный покой теломеразная актив ность падает. Это накладывает огра ничения на значение теломеразной активности как способа мониторинга за развитием опухоли в процессе лече ния.
Отсутствие активности или слабая теломеразная активность являются хо рошими диагностическими признака ми. Вероятно, часть случаев спонтан ного исчезновения опухолей связана с отсутствием в них теломеразной ак тивности.
решить эту проблему. Последствия воздействия ингибиторов теломера зы на стволовые клетки трудно про гнозировать. С одной стороны, клетки с имеющейся теломеразой способны восстанавливать свои те ломеры после отмены ингибиторов теломеразы. С другой стороны, из вестно, что стволовые клетки хотя и обладают теломеразной активно стью, однако эта активность ком пенсирует укорачивание теломер не полностью. Насколько нам извест но, деление стволовых клеток — от носительно редкое событие, поэтому ограниченное во времени лечение не должно серьезно сказаться на стволовых клетках.
Можно ожидать, что лечение инги биторами теломеразы вызовет уменьшение пролиферативного по тенциала стволовых клеток, что мо жет выразиться в их преждевремен ном старении.
Как мы уже отмечали, в опухоле вых клетках человека обычно отсутст вуют (не работают) механизмы нор-
188
мального пролиферативного старения. |
3 . 5 . 3 . 3 . Возможные способы |
|
Действие ингибитора теломеразы вы |
воздействия на опухоль, |
|
зовет укорачивание теломер, которое |
опосредованные теломеразой |
|
не будет индуцировать остановки про |
и теломерами |
|
лиферации клеток. |
|
|
|
|
3.5.3.3.1. Вакцинация |
Чрезвычайное укорачивание тело- |
или генерация цитотоксических |
|
мер приведет к потере теломерами |
Т-лимфоцитов |
|
функции |
защиты от теломерных |
|
слияний. |
Это вызовет множест |
При функционировании теломера |
венные изменения кариотипа, что зы в клетке фрагменты теломеразной
будет |
способствовать |
быстрому |
обратной |
транскриптазы |
экспониру |
||||||||||||||||||
появлению |
|
устойчивых |
|
к |
пред |
ются на клеточной поверхности и мо |
|||||||||||||||||
ложенному |
|
методу |
|
лечения |
кле |
гут служить мишенью иммунного от |
|||||||||||||||||
ток. |
|
|
|
|
|
|
|
|
|
|
|
вета. Выделен ряд пептидов, которые |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
связываются |
с |
антигенами |
главного |
|||||||
Подавляющая масса |
клеток |
поте |
комплекса гистосовместимости 1-го |
||||||||||||||||||||
ряет теломеры полностью (на от |
класса (HLA-A2.1), представленными |
||||||||||||||||||||||
дельных |
хромосомах), |
что |
повлечет |
на поверхности значительной доли Т- |
|||||||||||||||||||
их гибель. В зависимости от устой |
лимфоцитов. В модельных системах |
||||||||||||||||||||||
чивости клеток к апоптозу будут на |
наблюдали |
значительное |
цитотокси- |
||||||||||||||||||||
блюдаться |
|
различные |
сочетания |
ческое |
действие. |
Эффективность |
и |
||||||||||||||||
апоптоза и некроза. Повышение ге |
специфичность |
подобной |
процедуры |
||||||||||||||||||||
нетической |
|
изменчивости |
|
клеток |
пока не ясны. |
|
|
|
|
|
|
|
|||||||||||
будет |
способствовать |
появлению |
как |
|
|
|
|
|
|
|
|
|
|
|
|||||||||
резистентных |
|
к |
препарату |
клеток, |
3.5.3.3.2. Ингибиторы |
обратных |
|
||||||||||||||||
так и |
клеток, |
способных |
к длитель |
|
|||||||||||||||||||
транскриптаз |
|
|
|
|
|
|
|
||||||||||||||||
ному |
переживанию |
|
в |
состоянии |
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
пролиферативного покоя. Все это |
Поскольку теломераза относится |
к |
|||||||||||||||||||||
должно |
ограничить применение |
ин |
обратным |
транскриптазам |
и |
характе |
|||||||||||||||||
гибиторов |
теломеразы. |
|
|
|
|
|
ризуется малой разборчивостью к суб |
||||||||||||||||
Другим |
серьезным |
недостатком |
стратам |
|
(сравнительно |
|
с |
другими |
|||||||||||||||
антителомеразной |
терапии |
является |
клеточными полимеразами), |
возмож |
|||||||||||||||||||
относительно |
|
большой |
латентный |
но применение различных модифи |
|||||||||||||||||||
период от момента начала лечения |
цированных |
нуклеозидов |
с |
целью |
|||||||||||||||||||
до наступления эффекта. Опыты по |
терминировать |
теломеразный |
син |
||||||||||||||||||||
подавлению |
теломеразы |
в |
культуре |
тез. Наиболее известны попытки по |
|||||||||||||||||||
клеток |
почти |
однозначно |
подтвер |
давить функцию теломеразы с помо |
|||||||||||||||||||
ждают это. Постоянная экспрессия |
щью азидотимидина. Еще предстоит |
||||||||||||||||||||||
антисмысловой конструкции против |
выяснить, |
|
насколько |
эффективной |
|||||||||||||||||||
Р Н К компонента теломеразы приво |
может оказаться такая терапия. Ос |
||||||||||||||||||||||
дит к гибели клетки HeLa примерно |
новной |
проблемой |
является |
токсич |
|||||||||||||||||||
через 25 удвоений популяции (УП). |
ность нуклеозидов, связанная с воз |
||||||||||||||||||||||
Такая же процедура, проведенная с |
действием на митохондриальную по- |
||||||||||||||||||||||
клетками |
злокачественной |
глиомы, |
лимеразу. |
|
|
|
|
|
|
|
|
|
|||||||||||
приводит к частичной дифференци - |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
ровке или апоптозу через 30 УП. Ка- |
3.5.3.3.3. Олигонуклеотидные |
|
|
||||||||||||||||||||
техины |
зеленого |
чая, |
которые |
по |
|
|
|||||||||||||||||
стратегии |
|
|
|
|
|
|
|
||||||||||||||||
давляют |
теломеразную |
активность в |
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
опухолевых |
клетках |
человека |
U-937 |
Олигонуклеотидные |
стратегии |
яв |
|||||||||||||||||
иНТ-29, в культуре вызывали их ляются самыми специфичными и ма
старение через 35 и 60 УП соответ |
лотоксичными. Основная проблема — |
ственно. |
доставка олигонуклеотидов в клетки. |
189
Введение в клетки гена, кодирую |
ходит |
сборка |
дефектной |
теломеразы, |
||||||||||||||||||
щего |
антисмысловую |
последователь |
что ведет к подавлению теломеразной |
|||||||||||||||||||
ность к РНК компоненту теломеразы, |
активности, |
укорачиванию теломер и |
||||||||||||||||||||
приводит |
к подавлению теломеразной |
индукции апоптоза. Недостаток — не |
||||||||||||||||||||
активности и к апоптозу большинства |
возможность введения гена во все |
|||||||||||||||||||||
клеток. Недостаток |
— |
невозможность |
опухолевые |
клетки. |
|
|
|
|||||||||||||||
введения гена во все опухолевые |
До сих пор не найдено эффектив |
|||||||||||||||||||||
клетки. |
|
|
|
|
|
|
|
|
|
|
|
ных |
регуляторов |
экспрессии |
компо |
|||||||
Введение |
|
в |
клетки |
олигонуклеоти |
нентов теломеразы и веществ, блоки |
|||||||||||||||||
дов, |
комплементарных |
|
матрице |
|
РНК |
рующих сборку |
теломеразы. |
Поиски |
||||||||||||||
компонента теломеразы, ведет к по |
таких веществ являются, вероятно, са |
|||||||||||||||||||||
давлению |
теломеразной активности, |
мым перспективным направлением. |
||||||||||||||||||||
укорачиванию |
|
теломер |
и |
апоптозу. |
|
|
|
|
|
|
|
|
|
|||||||||
Применяют |
|
олигонуклеотиды |
с |
фос- |
3.5.3.3.5. Воздействие |
|
|
|||||||||||||||
форотиоатными |
связями, |
пептидные |
|
|
||||||||||||||||||
на теломеры |
|
|
|
|
|
|||||||||||||||||
нуклеиновые |
|
кислоты, |
2'-О-метил- |
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||||
Р Н К |
и |
2'-метоксиэтокси-РНК. |
Эти |
Воздействие |
на теломеры способно |
|||||||||||||||||
соединения |
|
характеризуются |
повы |
сделать их малодоступными для тело |
||||||||||||||||||
шенной устойчивостью к нуклеазам и |
меразы. Препараты, стабилизирующие |
|||||||||||||||||||||
повышенной |
специфичностью за счет |
квадруплексы, |
обладают |
антителоме- |
||||||||||||||||||
повышения |
|
температуры |
плавления |
разным действием, однако такие пре |
||||||||||||||||||
дуплексов. |
Для |
введения |
препаратов |
параты очень токсичны. Возможна |
||||||||||||||||||
используют |
различные |
методы |
транс |
специфическая |
доставка таких препа |
|||||||||||||||||
фекции и пероральное введение. Без |
ратов в теломеры с помощью теломе |
|||||||||||||||||||||
трансфекции |
эффективность |
метода |
разы. Если препарат, с одной сторо |
|||||||||||||||||||
низка. |
|
|
|
|
|
|
|
|
|
|
|
|
ны, будет субстратом теломеразы, а с |
|||||||||
Специфическое |
разрушение |
|
РНК |
другой — содержать группировку, ста |
||||||||||||||||||
компонента теломеразы — олигодезок- |
билизирующую квадруплексы |
(напри |
||||||||||||||||||||
сирибонуклеотид с присоединенной к |
мер, |
порфирин), |
токсичность таких |
|||||||||||||||||||
нему 2'-,5'-олигоаденилатной группой. |
препаратов резко понизится. |
|
||||||||||||||||||||
После |
узнавания |
олигонуклеотидом |
Гипотетически |
рассматриваются |
||||||||||||||||||
своей мишени 2'-,5'-олигоаденилат |
возможности |
неспецифического по |
||||||||||||||||||||
активирует |
эндогенные |
эндорибонук- |
вреждения теломер для |
избирательно |
||||||||||||||||||
леазы РНКазу L и РНКазу Н, которые |
го подавления опухолевого роста. По |
|||||||||||||||||||||
атакуют однонитевую |
Р Н К мишень. |
скольку теломеры |
репарируются хуже, |
|||||||||||||||||||
Олигонуклеотид |
может |
быть со |
чем любые другие области генома, а |
|||||||||||||||||||
единен с рибозимом, обладающим эн- |
опухолевые |
клетки в |
подавляющем |
|||||||||||||||||||
дорибонуклеазной |
активностью |
(так |
большинстве содержат очень короткие |
|||||||||||||||||||
называемый |
|
|
молоткоголовый |
|
рибо- |
теломеры по сравнению с окружаю |
||||||||||||||||
зим). После присоединения к мишени |
щей их нормальной тканью, то любой |
|||||||||||||||||||||
рибозим |
разрушает ее. |
|
|
|
|
окислительный |
стресс |
вызовет более |
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
скорую гибель |
опухолевых |
клеток. |
||||||
3.5.3.3.4. Воздействие |
|
|
|
|
|
|
|
* * * |
|
|
|
|||||||||||
на экспрессию генов теломеразы |
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
||||||||||||||
Введение гена доминантно-нега |
Поскольку |
реактивация теломера |
||||||||||||||||||||
тивного |
мутанта |
теломеразной |
об |
зы является |
|
наиболее |
универсальной |
|||||||||||||||
ратной |
транскриптазы |
в |
эксперимен |
характеристикой |
опухолевых |
клеток |
||||||||||||||||
тах |
является |
наиболее |
эффективным |
(85 % ) , измерение уровня теломераз |
||||||||||||||||||
способом |
|
|
подавления |
теломеразы. |
ной активности и подавление теломе |
|||||||||||||||||
При введении такого гена его продукт |
разы может и должно применяться в |
|||||||||||||||||||||
конкурирует за |
|
Р Н К компонент тело |
терапии опухолевых заболеваний. |
|||||||||||||||||||
меразы с |
нормальным |
геном. Проис |
Подавление функции теломеразы в |
|||||||||||||||||||
190
опухолевых клетках человека ведет к укорачиванию их теломер и вызывает состояние кризиса, но не старения. Явление кризиса сопровождается од новременно идущими процессами ги бели клеток и пролиферации. Много кратно возрастает генетическая измен чивость клеток, что вызывает быстрое появление резистентных форм. Для изучения действия ингибиторов тело меразы не подходят модели с исполь зованием животных, поскольку у чело века — особая регуляция теломеразы.
Рекомендуемая литература
Егоров Е. Е. Теломераза, старение, рак // Мол. биол. - 1997. - Т. 31. - С. 16.
Егоров Е. Е. Вокруг теломеразы // Мол. биол. - 1999. - Т. 33. - С. 385-392.
Егоров Е. Е. Теломеры, теломерная Д Н К , хромосомы // Биол. мембраны. — 2001. - Т. 18. - С. 240-247.
Griffith J. D. et al. Mammalian telomeres end in a large duplex loop // Cell. — 1999. — Vol. 97. - P. 503-514.
Harley С. B. et al. Telomerase, cell immortali ty and cancer // Cold Spring Harbor Sym posia on Quantitative Biology. — 1994. — Vol. 59. - P. 307-315 .
Wright W. E., Shay J. W. The two-stage mechanism controlling cellular senescence and immortalization // Exp. Gerontol. — 1992. - Vol. 27. - P. 383-389.
3.6. Эпигенетические изменения в опухолевых клетках. Роль метилирования ДНК в канцерогенезе
Н.П. Киселева,
А.В. Лихтенштейн
Вопрос о соотношении роли гене тических и эпигенетических измене ний в клетках, приводящих к развитию опухоли, десятилетиями являлся пред метом дискуссий. В настоящее время не вызывает сомнений огромная роль, которую играют в канцерогенезе гене тические изменения, т. е. изменения в структуре генов. Наибольшие успехи в этом вопросе достигнуты в отноше нии таких ключевых для опухолевого роста генов, как онкогены, опухоле
вые супрессоры, гены системы репа рации ДНК, гены, контролирующие апоптоз, рассмотренные в предыдущих главах. На фоне этих успехов роль эпи генетических изменений, т. е. измене ний, не затрагивающих генетических потенций генов, но приводящих к на следуемой модификации их экспрес сии, отодвигалась на второй план. За последние годы, однако, накопился экспериментальный материал, свиде тельствующий о том, что не менее важную роль в канцерогенезе могут играть эпигенетические изменения, влияющие на экспрессию многих клю чевых для канцерогенеза генов, в том числе и перечисленных выше. Более того, появились экспериментальные данные, которые говорят о взаимодей ствии эпигенетических и генетических событий в процессе опухолевой про грессии, когда одни события провоци руют возникновение других.
Механизмы, контролирующие эти процессы, во многом остаются неяс ными. Ниже будут рассмотрены дан ные о нарушении закономерностей метилирования Д Н К в опухолевых клетках, которые могут приводить к изменению экспрессии генов, вовле ченных в канцерогенез, а также инду цировать генетические изменения.
Метилирование Д Н К — процесс энзиматического присоединения ме тильной группы к основаниям в со ставе Д Н К . По своей природе это эпигенетический процесс, так как при этом не происходит изменения гене тического кода. Метилирование Д Н К обнаружено у прокариот и высших эу кариот. Наиболее сложные функции оно выполняет у млекопитающих; особенности этих функций и будут здесь рассмотрены.
Долгое время функции метилиро вания оставались непонятными, тем более что у некоторых эукариот (дрожжи) оно не было обнаружено. Благодаря значительному прогрессу в исследовании этой модификации Д Н К , достигнутому в последнее деся тилетие, обозначились многообразие и сложность функций метилирования.
191
Не изменяя генетического кода, мети лирование вмешивается в ДНК-бел ковые взаимодействия в хроматине, благодаря чему принимает участие в структурной и функциональной орга низации генома. Оно вовлечено в та кие фундаментальные процессы жиз недеятельности клетки, как регуляция экспрессии генов и поддержание це лости генома. Очевидно, что многооб разие функций метилирования и важ ность процессов, в которых оно участ вует, предполагает наличие тонкой и жесткой регуляции его. В исследова ниях на экспериментальных моделях было показано, что нарушение этой регуляции в эмбриогенезе может при водить к гибели организма. Наруше ние метилирования в соматических клетках взрослого организма наблю дается при некоторых заболеваниях человека, в том числе и при злокаче ственных новообразованиях.
В данном разделе рассмотрены со временные представления о метили ровании Д Н К и его роли в канцероге незе, а также о возможности исполь зования этих знаний в фундаменталь ной и практической онкологии.
3.6.7. Роль метилирования ДНК в нормальных клетках
Для понимания роли нарушения метилирования при канцерогенезе не обходимо знание закономерностей этого процесса в нормальном орга низме.
Метилирование не наследуется от родительских гамет, а устанавливается заново в процессе эмбриогенеза. Это многостадийный процесс, в ходе ко торого в результате волн метилирова ния и деметилирования, сменяющих друг друга в строго определенные мо менты эмбриогенеза, достигается ха рактерный для данного типа клеток паттерн метилирования Д Н К , кото рый затем поддерживается неизмен ным при делении клеток взрослого организма. У млекопитающих метили рование осуществляется ферментатив ным путем по пятому положению
кольца цитозинового остатка в первые минуты после репликации ДНК . Мо дификации могут подвергаться не все цитозиновые остатки, а только те, ко торые образуют динуклеотид 5'CpG. Цитозиновый остаток в составе 5'GрС-динуклеотида или любых дру гих нуклеотидов не метилируется. По разным оценкам 70—80 % CpG-ди- нуклеотидов в геноме метилированы, а остальные не имеют этой модифика ции. Различают два типа метилирова ния: метилирование de novo и поддер живающее. Метилирование de novo осуществляется в эмбриогенезе в сай тах, содержащих CpG-динуклеотиды, которые не метилированы в обеих комплементарных цепях ДНК . Под держивающее метилирование осуще ствляется только в тех сайтах вновь синтезированной цепи ДНК, где в ро дительской цепи уже содержится CpG-динуклеотид с метилированным остатком цитозина. Для клеток взрос лого организма характерно только поддерживающее метилирование, что и обеспечивает сохранение относи тельно неизменным паттерна метили рования ДНК, присущего данному ти пу клеток, при их делении.
В ходе эволюции CpG-динуклеоти ды прогрессивно элиминировались из генома, и сейчас содержание их со ставляет от 5 до 10 % расчетной вели чины встречаемости у человека и мы ши. Предполагается, что именно ме тилирование сыграло критическую роль в этом процессе, так как извест но, что 5-метилцитозин легко дезаминируется и превращается в тимин. Сохранившиеся к настоящему време ни CpG-динуклеотиды распределены по геному неравномерно. В геноме су ществуют короткие (от 0,5 до 3 т. п.н.) последовательности, где CpG-динук леотиды распределены кластерами, плотность их близка к расчетной, а содержание G + C превышает 60 %. Такие последовательности получили название CpG-островков. В остальной части Д Н К CpG-динуклеотиды рас пределены равномерно и редко. Боль шая часть CpG-островков расположе-
192
на в 5' регуляторных районах генов, и примерно половина генов человека и мыши имеет CpG-островки. К их чис лу относятся так называемые гены до машнего хозяйства, которые экспрессируются во всех тканях, некоторые тканеспецифичные гены, многие про тоонкогены и гены-супрессоры опухо левого роста. У второй части генов, в том числе у большинства тканеспецифичных генов, регуляторные зоны не имеют CpG-островков, хотя и содер жат индивидуальные CpG-динуклео тиды примерно с такой же плотно стью, как и смысловые последователь ности гена. Для этих двух типов регу ляторных зон, различных по распре делению CpG-динуклеотидовов, мети лирование устанавливается в эмбрио генезе по-разному.
CpG-островки, ассоциированные с регуляторными зонами генов, не ме тилированы во всех тканях эмбриона
и взрослого организма, в том числе и
вгаметах, независимо от того, экспрессируются в них гены или нет. Это неотъемлемое свойство CpG-остров ков — отсутствие метилирования во всех тканях на всех стадиях развития, по-видимому, способствовало сохра нению нормальной частоты встречае мости CpG-динуклеотидов в них бла годаря большей стабильности цитозина по сравнению с 5-метилцитозином. Механизм, позволяющий CpG-ост-
ровкам |
избегать метилирования de |
novo в |
эмбриогенезе, пока не извес |
тен. Предполагают, что защитную роль в этом процессе играют специ фические нуклеотидные последова тельности. В настоящее время защи щающая от метилирования de novo роль доказана по крайней мере для одной последовательности, являю щейся одновременно сайтом узнава ния транскрипционного фактора Spl. В силу того что CpG-островки в регу ляторных зонах не метилированы не зависимо от экспрессии генов, можно сказать, что метилирование не участ вует в регуляции экспрессии этих ге нов в нормальных клетках, которая регулируется другими механизмами.
Исключение из этого правила состав ляют только гены, расположенные на инактивированной Х-хромосоме у са мок, а также импринтированные ге ны, которые транскрибируются толь ко с одного из двух аллелей (материн ского или отцовского). У этих генов CpG-островки метилированы в нор мальных клетках. Кроме того, в неоплазиях человека и животных на блюдается аберрантное метилирова ние CpG-островков ряда генов (будет рассмотрено ниже). Во всех этих ис ключениях метилирование CpG-ост ровков сопровождается подавлением транскрипции генов, наследуемым при делении клетки, т. е. для этих ге нов существует корреляция между ме тилированием и долговременным по давлением транскрипции.
В отношении группы тканеспецифичных генов, лишенных CpG-ост ровков, было высказано предположе ние, согласно которому они подверга ются метилированию de novo во время имплантации эмбриона. Затем во вре мя дифференцировки CpG-сайты в их регуляторных зонах деметилируются только в тех тканях, где они экспрессируются во взрослом организме, од нако убедительные доказательства в пользу этой гипотезы пока отсутству ют. С развитием методов, позволяю щих анализировать статус метилиро вания каждого остатка цитозина, бы ло показано, что регуляторные рай оны ряда генов также слабо метили рованы в некоторых тканях, где они не экспрессируются, как и в тканях, где есть их экспрессия. В последнее время удалось показать также, что ис кусственно вызванное у мышиного эмбриона деметилирование Д Н К не приводит к несвоевременной экспрес сии исследованных тканеспецифичных генов или к экспрессии их не в тех типах тканей, в которых они должны экспрессироваться. Возмож но, что деметилирование регулятор ных районов этих генов является не обходимым, но недостаточным усло вием для экспрессии; не исключено также, что метилирование не играет
13-7908 Д. Г. Заридзе |
193 |
существенной роли в регуляции экс |
этот механизм не может быть универ |
||||||||||||||||||||||
прессии этой группы генов ни в эм |
сальным, так как многие другие фак |
||||||||||||||||||||||
бриогенезе, ни во взрослом орга |
торы |
|
транскрипции |
нечувствительны |
|||||||||||||||||||
низме. |
|
|
|
|
|
|
|
|
к метилированию их сайтов связыва |
||||||||||||||
К |
числу последовательностей, |
ко |
ния или узнают сайты, не содержащие |
||||||||||||||||||||
торые |
обогащены |
по |
содержанию |
CpG-динуклеотидов. Благодаря иссле |
|||||||||||||||||||
CpG-динуклеотидов, относятся также |
дованиям |
|
последнего |
десятилетия |
|||||||||||||||||||
транспозоны, которые |
составляют не |
сформировалось представление о вто |
|||||||||||||||||||||
менее 25 % генома человека. Эти па |
ром |
|
механизме |
подавления |
транс |
||||||||||||||||||
разитические элементы метилированы |
крипции, |
|
который |
обладает |
универ |
||||||||||||||||||
во всех изученных сайтах генома |
сальными возможностями. Само по |
||||||||||||||||||||||
взрослого организма. Многие транс |
себе наличие метилцитозина в ДНК |
||||||||||||||||||||||
позоны |
имеют |
сильные |
промоторы, |
не препятствует продвижению РНК- |
|||||||||||||||||||
способные |
поддерживать |
эффектив |
полимеразы, |
однако |
оно |
|
изменяет |
||||||||||||||||
ную |
транскрипцию. |
Метилирование |
структуру хроматина, делая ее недос |
||||||||||||||||||||
их коррелирует с подавлением транс |
тупной для |
формирования |
комплекса |
||||||||||||||||||||
крипционной активности, что способ |
белков, |
инициирующих |
|
транскрип |
|||||||||||||||||||
ствует защите генома от их дальней |
цию. Установлено, что неметилиро- |
||||||||||||||||||||||
шего |
распростанения, |
для |
которого |
ванное состояние CpG-островков ас |
|||||||||||||||||||
необходимо образование новых копий |
социировано |
с открытой |
конфигура |
||||||||||||||||||||
Д Н К транспозонов на |
матрице РНК . |
цией хроматина, в которой гистоны, |
|||||||||||||||||||||
Кроме того, метилированы во всех |
формирующие |
нуклеосому, |
находятся |
||||||||||||||||||||
изученных |
сайтах |
взрослого |
организ |
в ацетилированной форме (транс- |
|||||||||||||||||||
ма сателлитные повторы. Эти парази |
крипционно-компетентный |
|
хрома |
||||||||||||||||||||
тические |
некодирующие |
последова |
тин). Метилирование CpG-сайтов |
||||||||||||||||||||
тельности, составляющие около 10 % |
приводит |
к формированию репресси |
|||||||||||||||||||||
генома, по-видимому, играют важную |
рующего |
многокомпонентного белко |
|||||||||||||||||||||
роль в поддержании структурной ор |
вого |
комплекса, |
который |
индуцирует |
|||||||||||||||||||
ганизации хромосом, а их метилиро |
деацетилирование |
гистонов. |
|
Послед |
|||||||||||||||||||
вание, очевидно, выполняет также за |
нее в свою очередь определяет транс- |
||||||||||||||||||||||
щитные функции (см. ниже). |
|
|
крипционно-неактивную |
|
|
структуру |
|||||||||||||||||
Таким |
образом, |
метилирование |
хроматина. |
Основой |
для |
формирова |
|||||||||||||||||
ния |
такого |
комплекса |
служат |
метил- |
|||||||||||||||||||
принимает |
участие |
в |
регуляции |
экс |
|||||||||||||||||||
цитозинсвязывающие |
белки. |
Наибо |
|||||||||||||||||||||
прессии |
генов у млекопитающих, |
вы |
|||||||||||||||||||||
лее изученный из |
них |
МеСР2 облада |
|||||||||||||||||||||
полняя специфические функции, свя |
|||||||||||||||||||||||
ет способностью связываться только с |
|||||||||||||||||||||||
занные |
с |
аллельспецифической |
экс |
||||||||||||||||||||
метилированными |
CpG-динуклеоти- |
||||||||||||||||||||||
прессией |
(импринтинг, |
инактивация |
|||||||||||||||||||||
дами, |
локализуется в ядре |
в |
неактив |
||||||||||||||||||||
Х-хромосомы), |
подавлением |
экспрес |
|||||||||||||||||||||
ном |
хроматине в |
комплексе |
с гисто- |
||||||||||||||||||||
сии |
транспозонов |
и, |
возможно, |
тка- |
|||||||||||||||||||
новыми деацетилазами. Связываясь с |
|||||||||||||||||||||||
неспецифичных генов, |
а также в слу |
||||||||||||||||||||||
метилированным |
|
участком |
ДНК, |
||||||||||||||||||||
чаях |
аберрантного |
метилирования |
|
||||||||||||||||||||
МеСР2 через |
корепрессор |
взаимодей |
|||||||||||||||||||||
CpG-островков при некоторых болез |
|||||||||||||||||||||||
ствует |
с |
деацетилазами и |
индуцирует |
||||||||||||||||||||
нях. |
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
подавление |
транскрипции |
путем де- |
||||||||||||
Метилирование |
подавляет |
экс |
|||||||||||||||||||||
ацетилирования гистонов |
|
и |
модифи |
||||||||||||||||||||
прессию |
генов |
на |
уровне транскрип |
|
|||||||||||||||||||
кации |
структуры |
хроматина. |
Таким |
||||||||||||||||||||
ции. Существует по крайней |
мере два |
||||||||||||||||||||||
образом, наряду с формированием ре |
|||||||||||||||||||||||
механизма, с помощью которых мети |
|||||||||||||||||||||||
прессирующего комплекса |
на |
основе |
|||||||||||||||||||||
лирование |
может |
препятствовать |
|||||||||||||||||||||
обычных |
белков-репрессоров, |
узнаю |
|||||||||||||||||||||
транскрипции. Один из них |
обуслов |
||||||||||||||||||||||
щих |
специфические |
последовательно |
|||||||||||||||||||||
лен |
неспособностью |
некоторых |
из |
||||||||||||||||||||
сти, |
высшие |
эукариоты |
с |
усложнен |
|||||||||||||||||||
вестных |
факторов |
транскрипции |
свя |
||||||||||||||||||||
ным |
геномом |
обладают дополнитель |
|||||||||||||||||||||
зываться с их сайтами узнавания, если |
|||||||||||||||||||||||
ным |
|
уникальным |
эпигенетическим |
||||||||||||||||||||
они |
метилированы. |
Очевидно, |
что |
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
194
механизмом для установления локаль ного деацетилироваия гистонов и соз дания неактивного состояния хрома тина, которое наследуется дочерними клетками при делении.
Метилирование цитозинового ос татка в Д Н К осуществляется фермен тативным путем. У млекопитающих, включая человека, известны четыре ДНК-(цитозин-5)-метилтрансферазы:
Dnmtl, Dnmt2, Dnmt3a и Dnmt3b. На основании серии биохимических экс периментов, в которых изучали спо собность ферментов катализировать метилирование различных ДНК-мат риц, а также серии генетических экс периментов с мышиными эмбрио нальными стволовыми клетками ES и мышами, у которых эти гены были инактивированы методом нокаута по отдельности или в разных сочетаниях, удалось установить свойства фермен тов. Dnmtl является главным претен дентом на роль фермента, осуществ ляющего поддерживающее метилиро вание in vivo. В делящихся клетках в культуре фермент локализуется в фо
кусах |
репликации |
Д |
Н К , его |
актив |
ность |
возрастает |
во |
время |
синтеза |
ДНК. В эмбрионах, лишенных генов двух других ферментов (dnmt3a и dnmt3b), Dnmtl не способен осущест влять метилирование de novo. In vitro этот фермент способен катализиро вать как поддерживающее метилиро вание, так и метилирование de novo, обладая при этом более высоким сродством к полуметилированной, чем к полностью деметилированной, ДНК-матрице, однако удаление N- концевого домена фермента стимули рует его de novo-метилирующую ак тивность.
Возможно, что in vivo активность и специфичность фермента по отноше нию к ДНК-матрице регулируются взаимодействиями N-концевого доме на с другими белками.
Предполагают, что нарушение функций именно этого фермента при канцерогенезе вносит вклад в абер рантное метилирование CpG-остров ков (см. ниже).
13*
Dnmt3a и Dnmt3b необходимы для метилирования de novo. Они экспрессируются на высоком уровне в эм брионах и на очень низком уровне в соматических клетках взрослой мыши. Инактивация этих генов в ES-клетках или в ранних эмбрионах блокирует метилирование de novo, но не влияет на поддерживающее метилирование введенных в клетки экзогенных мети лированных последовательностей. Повидимому, эти ферменты различаются по своим функциям, так как Dnmt3b необходима для метилирования центромерных минорных сателлитных повторов, a Dnmt3a — нет. Функции Dnmt2 остаются пока не ясными, так как инактивация этого гена в клетках ES не изменяет ни того, ни другого вида метилирования.
Вопрос о том, как регулируется ме тилирование de novo и как оно соот носится с поддерживающим метили рованием, важен не только для эм бриогенеза, но и для канцерогенеза, так как в опухолевых клетках наблю дается глобальное нарушение паттер на метилирования Д Н К . Механизм формирования сложного паттерна ме тилирования генома в эмбриогенезе остается во многом неясным. Одна из гипотез предполагает существование динамического равновесия между по стоянно действующими и противо стоящими друг другу процессами экс пансии метилирования и защиты от него. Гипотеза возникла как результат анализа метилирования 5'-района ге на домашнего хозяйства — аденинфосфорибозилтрансферазы. На опре деленной стадии эмбрионального раз вития фермент, способный к реакции de novo, метилирует последователь ность, состоящую из повторяющихся элементов и расположенную вблизи промотора этого гена — так называе мый центр метилирования. После этого метилирование распространяет ся по направлению к промотору, рас положенному в CpG-островке, по-ви димому, с помощью поддерживающей метилтрансферазы. Сайты связывания фактора SP1, блокирующие процесс,
195
защищают CpG-островок от экспан сии метилирования. В силу конкурен ции между импульсами метилирова ния, исходящими из центра метили рования, и защищающими структура ми промотора промежуточные после довательности находятся в динамиче ски нестабильном состоянии и содер жат полуметилированные CpG-динук леотиды, т. е. такие, которые метили рованы лишь в части молекул ДНК . Важное следствие этой гипотезы — представление о возможности измене ния профиля метилирования в клет ках взрослого организма при каждом делении в определенных пределах. Изменение профиля может возник нуть при усилении или ослаблении одного из двух противоборствующих процессов на протяжении нескольких клеточных делений. Именно это и происходит, по-видимому, при старе нии и злокачественной трансформа ции клетки.
3.6.2. Роль деметилирования ДНК в нормальной клетке
Вскоре после оплодотворения большинство метилированных в гаме тах CpG-сайтов подвергается деметилированию. Это деметилированное состояние Д Н К сохраняется вплоть до имплантации эмбриона, когда боль шинство CpG-сайтов заново метили руются, за исключением тех, которые избегают метилирования в составе CpG-островков. Позднее в процессе дифференцировки происходит ло кальное деметилирование индивиду альных генов. В результате этих по следовательных событий 70—80 % CpG-сайтов в геноме человека и мы ши оказываются метилированными, в том числе сайты, расположенные в многочисленных повторяющихся по следовательностях и транспозонах. В настоящее время ничего неизвестно о том, как происходит глобальное деме тилирование в эмбриогенезе.
Что касается механизма локально го деметилирования, то для несколь ких тканеспецифичных генов установ
лено, что этот процесс контролирует ся специфическими нуклеотидными последовательностями, так называе мыми цис-действующими элементами. Например, в процессе дифференци ровки В-лимфоцитов гены иммуног лобулинов проходят ряд структурных перестроек. Этот процесс называется реаранжировкой V(D)J локуса имму ноглобулинов, которая осуществляет ся путем генетической рекомбинации V-, D- и J-фрагментов генов. На оп ределенной стадии развития В-клеток происходит специфическое деметили рование некоторых участков ДНК в этом локусе, которое контролируется несколькими цис-действующими эле ментами, расположенными в регуля торных зонах генов иммуноглобули нов. Эти элементы узнаются специ фическими транс-действующими бел ковыми факторами. Так же как и в случае элемента SP1, принимающего участие в защите CpG-островков от метилирования, один из элементов, направляющих деметилирование, ока зался сайтом связывания транскрип ционного фактора NF-кВ. Если ис кусственно удалить комплекс цис-дей- ствующих элементов, которые направ ляют деметилирование в этом локусе, деметилирование не происходит, и перестройка оказывается невозмож ной. Кроме того, было показано, что сам транскрипционный фактор необ ходим для процесса деметилирования. Таким образом, возможно, что одни и те же цис-действующие элементы Д Н К и транс-действующие белковые факторы используются независимо и для транскрипции, и для локального деметилирования. Эти эксперименты, кроме того, говорят о связи деметили рования и рекомбинационных про цессов.
Существуют и другие доказательст ва в пользу того, что метилирование играет определенную роль в подавле нии рекомбинационных процессов. Гипометилирование Д Н К в мышиных ES клетках, лишенных функциональ ного гена dnmtl, сопровождается воз растанием частоты мутаций, причи-
196
ной которых |
являются |
митотические |
генетической нестабильности в опухо |
|||||||||||||||||||||
рекомбинации. |
|
Гипометилирование |
левой клетке. |
|
|
|
|
|
|
|
|
|
||||||||||||
сателлитных |
|
последовательностей, |
Гиперметилирование |
|
CpG-остров |
|||||||||||||||||||
сконцентрированных |
|
в |
центромер- |
ков. Понимание функции |
гипермети |
|||||||||||||||||||
ном районе хромосомы 1 человека, |
лирования |
локальных |
|
зон |
Д Н К |
в |
||||||||||||||||||
вызванное обработкой В-клеток деме- |
трансформированных in vitro и опухо |
|||||||||||||||||||||||
тилирующим агентом, приводит к не |
левых клетках пришло тогда, когда в |
|||||||||||||||||||||||
стабильности центромерной области и |
ряде исследований удалось связать ги- |
|||||||||||||||||||||||
образованию |
хромосом |
с |
аномальной |
перметилированные |
участки |
Д Н К |
с |
|||||||||||||||||
структурой. Возможно, |
что поддержа |
конкретными |
генами. |
|
Гиперметили |
|||||||||||||||||||
ние большей части CpG-сайтов в ге |
рование промоторного района как ме |
|||||||||||||||||||||||
номе в метилированном состоянии — |
ханизм |
инактивации |
впервые |
было |
||||||||||||||||||||
это общий механизм сохранения це |
описано |
для |
гена |
ретинобластомы |
||||||||||||||||||||
лости генома и подавления дестабили |
(Rb), |
известного |
супрессора |
опухоле |
||||||||||||||||||||
зирующей роли |
огромного количества |
вого роста. У 10 % пациентов со спо |
||||||||||||||||||||||
повторяющихся |
последовательностей |
радической |
формой |
ретинобластомы |
||||||||||||||||||||
и транспозонов у млекопитающих. |
|
наблюдалось |
гиперметилирование |
в |
||||||||||||||||||||
Таким |
образом, |
очевидно, |
что |
норме неметилированногО CpG-ост- |
||||||||||||||||||||
должен |
существовать |
|
жесткий |
кон |
ровка на 5'-конце гена Rb, которое |
|||||||||||||||||||
троль |
процесса деметилирования |
как |
коррелировало с отсутствием его экс |
|||||||||||||||||||||
в эмбриогенезе, так и в соматической |
прессии. Вследствие очень большого |
|||||||||||||||||||||||
клетке, нарушение которого приведет |
размера гена невозможно было ис |
|||||||||||||||||||||||
к нарушению точного баланса мети |
ключить |
наличия |
инактивирующих |
|||||||||||||||||||||
лирования и деметилирования и на |
мутаций у этих больных. Прямое до |
|||||||||||||||||||||||
рушению структуры и экспрессии ге |
казательство |
инактивируюшей |
роли |
|||||||||||||||||||||
нома. Как будет показано ниже, на |
метилирования в этом случае было |
|||||||||||||||||||||||
рушение этого баланса является не |
получено в экспериментах по метили |
|||||||||||||||||||||||
отъемлемым |
свойством |
опухолевой |
рованию |
промоторного |
района |
гена |
||||||||||||||||||
клетки. |
|
|
|
|
|
|
|
|
|
|
Rb in vitro с последующим введением |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
его в клетки методом трансфекции, |
||||||||||||
3.6.3. |
Нарушения |
|
|
|
|
которое |
сопровождалось |
подавлением |
||||||||||||||||
|
|
|
|
транскрипции. |
Затем |
|
для |
многих |
||||||||||||||||
метилирования |
ДНК |
|
|
|
||||||||||||||||||||
|
|
классических |
опухолевых |
|
супрессо |
|||||||||||||||||||
при |
|
канцерогенезе |
|
|
|
|
||||||||||||||||||
|
|
|
|
ров, а также для генов, которые участ |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
Ненормальный |
паттерн |
метилиро |
вуют |
в |
репарации Д Н К , |
регуляции |
||||||||||||||||||
вания |
Д Н К |
давно |
был известен |
как |
апоптоза или ассоциированы с мета- |
|||||||||||||||||||
постоянно присутствующее, но |
мало |
стазированием, было |
обнаружено ги |
|||||||||||||||||||||
понятное изменение в неоплазиях че |
перметилирование CpG-островков в |
|||||||||||||||||||||||
ловека и животных. Во всех без ис |
различных |
типах |
опухолей |
|
человека, |
|||||||||||||||||||
ключения |
исследованных |
неопласти |
всегда |
сопровождавшееся |
инактива |
|||||||||||||||||||
ческих |
|
клетках |
наблюдается |
дисба |
цией этих генов. Примеры таких ге |
|||||||||||||||||||
ланс метилирования. Он выражается, |
нов представлены в табл. 3.18. |
|
|
|||||||||||||||||||||
с одной стороны, в широко распро |
Наиболее |
широко |
|
распространен |
||||||||||||||||||||
страненном по геному деметилирова- |
ной (встречающейся во многих типах |
|||||||||||||||||||||||
нии |
нормально |
|
метилированных |
опухолей в большом проценте случа |
||||||||||||||||||||
CpG-сайтов и, с другой — в локаль |
ев) является инактивация путем ги |
|||||||||||||||||||||||
ном гиперметилировании CpG-ост |
перметилирования промоторного рай |
|||||||||||||||||||||||
ровков Д Н К . В свете описанных выше |
она гена ингибитора циклинзависи |
|||||||||||||||||||||||
функций |
метилирования |
в нормаль |
мых киназ р16INK4А. Функциональная |
|||||||||||||||||||||
ной клетке очевидно, что эти наруше |
потеря этого гена, участвующего в |
|||||||||||||||||||||||
ния могут изменять структуру хрома |
аресте клеточного цикла, является об |
|||||||||||||||||||||||
тина и функции Д Н К и вносить свой |
щей чертой многих типов опухолей. |
|||||||||||||||||||||||
вклад |
в |
создание |
фенотипической и |
Ранее |
для |
этого |
гена |
были |
описаны |
|||||||||||||||
197
Т а б л и ц а 3.18. Гены, для которых известно гиперметилирование CpG-островков в опухо
лях человека
Ген |
Опухоль |
|
Комментарии |
|
|
|
|
|
|
RB |
Спорадические |
рети |
Ген-супрессор опухолевого роста, инактивирую |
|
|
нобластомы |
|
щие мутации наблюдаются в семейных формах ре |
|
|
|
|
тинобластомы |
|
p15INK4B |
Лимфома Беркитта, |
Ген-супрессор опухолевого роста, потеря функций |
||
|
острые лейкозы |
|
нарушает сигнальный путь циклин D/Rb |
|
р16INK4А |
Многие солидные опу |
То же |
||
|
холи и лимфомы |
|
|
|
Е-кадхерин |
Мочевой пузырь, мо |
Ген-супрессор опухолевого роста, участвует в фор |
||
|
лочная железа, |
толстая |
мировании межклеточных контактов, потеря функ |
|
|
кишка, печень |
|
ций способствует инвазии и метастазированию |
|
h M L H 1 |
Толстая кишка |
|
Участвует в репарации Д Н К , потеря функций яв |
|
|
|
|
ляется причиной нестабильности микросателлит- |
|
|
|
|
ных повторов Д Н К |
|
HIC1 |
Почка, мозг, молочная |
Транскрипционный фактор, значение потери |
||
|
железа, толстая |
кишка |
функций неизвестно |
|
|
|
|
|
|
такие известные механизмы инактива ции в опухолевой клетке, как потеря одного аллеля в сочетании с сомати ческой мутацией оставшегося аллеля или гомозиготные делеции. В послед
нее время во многих опухолях обнару жены инактивация p16INK4A путем ги
перметилирования в обоих аллелях или сочетание гиперметилирования одного аллеля с соматической мутаци ей в другом. Для некоторых типов опухолей гиперметилирование являет
ся главным механизмом инактивации гена р16INK4A (для рака предстательной
железы, толстой кишки, мочевого пу зыря — от 60 до 90 % случаев). В ряде случаев из опухолей были получены клеточные линии, при обработке ко торых деметилирующим агентом на блюдалось деметилирование промо
торного района и восстановление транскрипции гена p16,INK4A. Такого
рода прямые доказательства инактивирующей роли гиперметилирования CpG-островков в промоторных рай онах были получены и для других ге нов, в том числе приведенных в табл. 3.18.
Стало очевидным, что аберрантное метилирование промоторного района является довольно распространенным
механизмом инактивации генов в опу холях. Для ряда опухолей было пока зано, что нарушение метилирования не ограничивается одним геном, а мо жет затрагивать одновременно не сколько известных генов, поврежде ние функций которых существенно для развития опухолей. В последнее время были разработаны методы, по зволяющие вести анализ CpG-остров ков отдельно от остальной части ДНК и одновременно дискриминировать метилированные и неметилированные CpG-островки. С помощью одного из таких методов было проведено мас штабное исследование аберрантного метилирования в 7 различных типах опухолей. В общей сложности было исследовано около 100 опухолей и в каждой из них более 1000 случайно отобранных CpG-островков, ассоциа ция которых с конкретными генами не была известна. По числу гиперметилированных CpG-островков иссле дованные типы опухолей разделились на две группы с относительно высо кой и низкой частотой метилирова ния. Внутри каждой группы индиви дуальные опухоли отличались друг от друга по этому признаку, но в гораздо меньшей степени, чем от представите-
198
лей других групп. Наряду с общими для нескольких типов опухолей гиперметилированными CpG-островками были обнаружены и такие, которые гиперметилированы только в одном типе, т. е. являются опухолеспецифичными. По оценкам, сделанным авторами, из 45 ООО CpG-островков, имеющихся в геноме человека, одно временно гиперметилированными мо гут оказаться в среднем 600 с доволь но широким разбросом — от 0 до 4500 в отдельных опухолях. Таким образом, дерегуляция метилирования CpG-ост ровков носит общий характер в опухо левых клетках. Возможно, что не все гиперметилированные CpG-островки из этого числа окажутся ассоцииро ванными с генами, вовлеченными в канцерогенез, и не все приведут к по давлению транскрипции генов, но и в этом случае очевидно, что аберрант ное метилирование 1—10 % CpG-ост ровков в клетке должно приводить к фенотипической нестабильности. По следняя в свою очередь может, по-ви димому, провоцировать генетическую нестабильность, как это было показа но для спорадических колоректальных опухолей. Причиной нестабильности микросателлитных повторов, харак терной для этих опухолей, в 70—80 % случаев является гиперметилирова ние промотора гена hMLH1, необхо димого для репарации неспаренных участков Д Н К . Кроме того, вновь воз никшие метилированные CpG-сайты могут служить основой для образова ния точечных мутаций. Метилирова ние цитозина приводит к нестабиль ности его аминогруппы и спонтанно му дезаминированию с образованием тимина, что имеет место даже при обычных условиях в результате тепло вых флюктуации. Возникающий де фект (неспаренные основания G—Т) индуцирует систему репарации. Тимин — естественное основание Д Н К (не распознается ферментами репара ции как нечто чужеродное), в связи с чем ситуация разрешается по-разно му: восстановлением исходной после довательности (пары G — С) или воз
никновением мутации (заменой пары G — С на А—Т). Хотя ферменты репа рации с большой частотой удаляют тимин, мутации тем не менее возни кают часто. Именно нестабильностью 5'-метилцитозина и многочисленными заменами пар G — С на А—Т, имевши ми место на протяжении эволюции, объясняется относительно низкое со держание динуклеотида CpG в геноме человека.
Онкологический аспект проблемы состоит в том, что гиперметилирова ние G—С-богатых последовательно стей приводит к возрастанию частоты мутаций. Так, примерно из 300 мута ций гена р53, зарегистрированных в различных опухолях человека, 25— 30 % относятся к мутациям описанно го типа.
Открытие альтернативного мутаци ям механизма инактивации генов по служило толчком для развития мето дов поиска новых генов, вовлеченных в процесс образования опухолей, ос нованных на разнице в статусе мети лирования CpG-островков в нормаль ной и опухолевой клетке. Представ ленный в табл. 3.18 ген HIC1 — пер вый пример нового кандидата на роль гена — опухолевого супрессора, кото рый был выявлен недавно благодаря его ассоциации с CpG-богатым рай оном, гиперметилированным в опухо лях нескольких типов. HIC1 является транскрипционным фактором и мо жет быть активирован продуктом гена
р53.
Описанный выше метод глобаль ного скрининга опухолей в поисках аберрантно-метилированных CpG - островков в ближайшее время, по-ви димому, увеличит число таких генов.
Аберрантное метилирование CpG - островков — это раннее событие в процессе возникновения опухоли. На
пример, гиперметилирование промо торного района гена p16INK4A при плос
коклеточном раке легкого было обна ружено уже в гиперплазиях. В некото рых случаях гиперметилирование уда ется обнаружить на более ранних ста диях опухолевой прогрессии, чем по-
199
тери |
гетерозиготности. |
Более |
того, |
типов |
|
мышиных |
и |
|
человеческих |
|||||||||||||||||
оказалось, что и в нормальных клет |
трансформированных |
|
клеток в |
куль |
||||||||||||||||||||||
ках имеет место локальное гипермети |
туре |
|
было |
показано |
|
многократное |
||||||||||||||||||||
лирование некоторых генов (эстроге- |
возрастание |
активности |
Dnmtl |
по |
||||||||||||||||||||||
новый рецептор ER, MyoD, IGF2). Ме |
сравнению |
с нормальными |
клетками, |
|||||||||||||||||||||||
тилирование промоторов этих генов — |
которое |
наблюдается |
одновременно с |
|||||||||||||||||||||||
феномен, связанный со старением (не |
возрастанием в несколько раз уровня |
|||||||||||||||||||||||||
выявляется у молодых, но обнаружи |
экспрессии м Р Н К . |
|
Известно, |
что |
||||||||||||||||||||||
вается и прогрессивно нарастает по |
фермент является пролиферативно-за- |
|||||||||||||||||||||||||
мере |
старения |
индивидуума). |
|
|
|
висимым и увеличение его экспрессии |
||||||||||||||||||||
Инактивируя |
гены, |
тормозящие |
в опухолях связано с увеличением |
|||||||||||||||||||||||
клеточный |
|
рост |
|
и |
стимулирующие |
числа делящихся клеток в них по |
||||||||||||||||||||
дифференцировку, |
этот |
процесс |
соз |
сравнению |
с нормальными |
тканями, |
||||||||||||||||||||
дает |
предпосылки |
к |
возникновению |
однако одним увеличением числа де |
||||||||||||||||||||||
опухоли. Во многих опухолях эти ге |
лящихся клеток нельзя объяснить де- |
|||||||||||||||||||||||||
ны часто метилированы. По всей ве |
регуляцию экспрессии Dnmtl в опу |
|||||||||||||||||||||||||
роятности, связанное с возрастом ме |
холях. |
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
тилирование генов в разных тканях |
На |
экспериментальных |
моделях |
|||||||||||||||||||||||
организма — обычное явление, пред |
были |
|
получены |
прямые |
доказательст |
|||||||||||||||||||||
располагающее к возникновению опу |
ва связи Dnmtl с процессом транс |
|||||||||||||||||||||||||
холи. |
|
|
|
|
|
|
|
|
|
|
|
|
|
формации клеток в культуре. Введе |
||||||||||||
Раннее |
появление |
гиперметилиро |
ние |
клонированного |
гена |
мышиной |
||||||||||||||||||||
вания при образовании опухолей в со |
dnmtl в мышиные фибробласты NIH |
|||||||||||||||||||||||||
четании с возможностью его обнару |
ЗТЗ вызывало их трансформацию. В |
|||||||||||||||||||||||||
жения |
таким |
высокочувствительным |
других |
экспериментах |
при |
введении |
||||||||||||||||||||
методом, как П Ц Р , в настоящее время |
методом |
трансфекции |
клонированно |
|||||||||||||||||||||||
используется |
для |
попыток |
разработки |
го гена человеческой DNMT1 в им- |
||||||||||||||||||||||
методов |
диагностики. |
|
|
|
|
|
|
мортализованные |
человеческие |
фиб |
||||||||||||||||
Обратимость |
инактивации |
генов |
робласты было обнаружено, что CpG- |
|||||||||||||||||||||||
путем |
гиперметилирования |
сущест |
островки, расположенные в промо- |
|||||||||||||||||||||||
венным образом отличает этот эпиге |
торных зонах нескольких генов, в том |
|||||||||||||||||||||||||
нетический |
механизм |
|
от |
генетиче |
числе и приведенных в табл. 3.18 Е- |
|||||||||||||||||||||
ских |
|
механизмов, |
повреждающих |
кадгерина и гена HIC1, подвергаются |
||||||||||||||||||||||
первичную |
|
структуру |
Д Н К . |
Это |
от |
аберрантному метилированию. В то |
||||||||||||||||||||
крывает |
возможности |
|
для |
попыток |
же время ряд CpG-островков, ассо |
|||||||||||||||||||||
применения |
деметилирующих |
аген |
циированных с другими генами, в том |
|||||||||||||||||||||||
тов |
как |
средств, |
ограничивающих |
числе с геном р16INK4A, не изменяли |
||||||||||||||||||||||
инактивацию |
генов, нарушение |
экс |
своего |
статуса |
метилирования, |
т. е. |
||||||||||||||||||||
прессии которых вносит вклад в кан |
были по-прежнему защищены от ме |
|||||||||||||||||||||||||
церогенез. |
|
|
|
|
|
|
|
|
|
|
|
тилирования de novo, несмотря на по |
||||||||||||||
Механизмы нарушения метилирова |
стоянную экспрессию DNMT1 . Таким |
|||||||||||||||||||||||||
образом, |
Dnmtl |
|
вовлечена |
в процесс |
||||||||||||||||||||||
ния |
при канцерогенезе. |
Механизмы |
|
|||||||||||||||||||||||
трансформации, |
|
а |
постоянная |
экс |
||||||||||||||||||||||
нарушения |
|
контроля |
метилирования |
|
||||||||||||||||||||||
|
прессия фермента действительно при |
|||||||||||||||||||||||||
и появления |
de |
|
novo-метилирующей |
|||||||||||||||||||||||
|
водит |
к аберрантному |
метилированию |
|||||||||||||||||||||||
активности |
|
в |
клетках |
взрослого |
орга |
|||||||||||||||||||||
|
CpG-островков, |
|
но не глобальному, а |
|||||||||||||||||||||||
низма при |
канцерогенезе |
только |
на |
|
||||||||||||||||||||||
избирательному. Очевидно, что одной |
||||||||||||||||||||||||||
чинают |
исследоваться. |
Относительно |
||||||||||||||||||||||||
несвоевременной |
экспрессией |
фер |
||||||||||||||||||||||||
давно было |
замечено, |
|
что |
трансфор |
||||||||||||||||||||||
|
мента |
невозможно объяснить актива |
||||||||||||||||||||||||
мированные |
и |
опухолевые |
клетки |
от |
||||||||||||||||||||||
цию способности к метилированию de |
||||||||||||||||||||||||||
личаются |
от |
нормальных |
повышен |
|||||||||||||||||||||||
novo, |
которая существенно ограничена |
|||||||||||||||||||||||||
ным |
уровнем |
экспрессии |
и |
активно |
||||||||||||||||||||||
у него в |
нормальных |
клетках. По-ви |
||||||||||||||||||||||||
сти |
D n m t l , |
наиболее |
|
изученной |
из |
|||||||||||||||||||||
|
димому, |
в |
трансформированных и |
|||||||||||||||||||||||
ДНК-метилтрансфераз. |
Для |
многих |
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
200
опухолевых клетках нарушаются и ме ханизм, регулирующий специфич ность фермента по отношению к ДНК-матрице, и механизм защиты CpG-островков от метилирования. Окончательный эффект — инактива ция конкретного гена — складывает ся, вероятно, в результате взаимодей ствия по крайней мере этих двух ме ханизмов.
В нормальных клетках активность Dnmtl увеличивается с началом син теза Д Н К . В связи с этим можно было предполагать, что активность фермен та контролируется внутриклеточными путями, передающими митогенные сигналы. Действительно, в ряде экс периментов на клеточных культурах была установлена связь между мети лированием Д Н К и теми путями пере дачи митогенных сигналов, компо нентом которых является белок Ras. Введение в мышиные адренокортикальные опухолевые клетки VI экзо
генного |
негативного |
регулятора |
Ras |
|
(Gap) |
приводило |
к |
существенному |
|
снижению уровня |
м Р Н К и активно |
|||
сти D n m t l , что |
сопровождалось |
ре |
||
версией к нормальному фенотипу. Введение в полученные таким обра зом ревертанты экзогенного H-ras восстанавливало трансформирован ный фенотип и повышало уровень экспрессии м Р Н К и активности фер мента. В других экспериментах с клет ками, трансформированными при по мощи конститутивной экспрессии c-fos (Fos — модулятор активности Ras, расположенный в цепи передачи сигнала после Ras), было показано, что ингибирование экспрессии Dnmtl или ее активности восстанавливает нормальный фенотип клеток, несмот ря на высокий уровень экспрессии экзогенного c-fos, т. е. препятствует проявлению его онкогенного потен циала.
Таким образом, установлено, что Dnmtl является мишенью действия онкобелков Ras и Fos. В настоящее время на модельных системах разра батываются способы применения ин гибиторов D n m t l , ограничивающих ее
активность, для восстановления нор мального фенотипа опухолевых кле ток. Роль в канцерогенезе недавно от крытых ДНК-метилтрансфераз Dnmt2, Dnmt3a и Dnmt3b остается неясной.
Можно заключить, что аберрант ное метилирование — это эпигенети ческий механизм, который вносит свой вклад в процесс образования опухоли, подавляя экспрессию генов, т. е. создавая фенотипическую неста бильность в опухолевой клетке, а так же в ряде случаев — основу для гене тической нестабильности.
3.6.4. Гипометилироеание ДНК при канцерогенезе
Одним из первых изменений мети лирования, обнаруженных в неопла стических клетках, было снижение общего содержания метильных групп в Д Н К , которое наблюдалось, несмот ря на присутствие в них гиперметилированных локальных зон. Прямые до казательства связи гипометилирования Д Н К с неопластическим процес сом были получены в экспериментах на грызунах, в которых с помощью специальных диет добивались истоще ния донора метильных групп S-адено- зилметионина. Такое истощение при водило к образованию опухолей пече ни, которому предшествовало гипометилирование Д Н К . Несмотря на ши роко распространенное по всему ге ному опухолевой клетки деметилиро вание CpG-сайтов и явную ассоциа цию этого явления с процессом обра зования опухолей, до сих пор не уда валось связать его с изменениями в экспрессии конкретных генов, как это было сделано в случае гиперме тилирования.
Очевидно, что гены, содержащие CpG-островки в своих регуляторных зонах, не могут быть мишенью деме тилирования, так как они деметилированы во всех тканях. Исключение из этого правила представляют импринтированные гены с моноаллельной экспрессией, у которых CpG-ост ровки метилированы на одном из ал-
201
лелей, и гены на инактивированной Х-хромосоме.
Действительно, для опухолей пече ни, почки (спорадические опухоли Вильмса) и шейки матки было обна ружено аберрантное деметилирование материнского аллеля гена инсулиноподобного фактора роста IGF2, сопро вождавшееся его биаллельной экс прессией (потеря импринтинга). По теря импринтинга и биаллельная экс прессия этого гена для опухолей Вильмса часто обнаруживаются в при легающих к опухоли условно нор мальных тканях, т. е. является ранним событием при развитии опухоли, од нако количество импринтированных генов и генов, расположенных на X- хромосоме, явно недостаточно, чтобы нарушением их экспресии объяснить роль глобального деметилирования ге нома в опухолевых клетках.
Наиболее вероятным последствием гипометилирования Д Н К при канце рогенезе может быть возникающая в результате нарушения паттерна мети лирования нестабильность генома.
Для нормальных клеток известно, что искусственно вызванное гипометилирование Д Н К увеличивает часто ту реаранжировок эндогенных ретровирусов и повторяющихся последова тельностей, частоту образования делеций и транслокаций некоторых уни кальных генов и является причиной хромосомных аномалий (см. выше). Для опухолевых клеток пока отсутст вуют экспериментальные данные, ко торые демонстрировали бы, что пере численные нарушения, всегда при сутствующие в них, являются пря мым следствием гипометилирования
ДН К .
Всвете дестабилизирующей роли гипометилирования Д Н К в нормаль ных клетках, однако, можно предпо лагать, что гипометилирование вносит свой вклад в создание мутаторного фенотипа и в опухолевой клетке. Дру гими словами, являясь эпигенетиче ской модификацией Д Н К , гипомети лирование, по-видимому, может при нимать участие в создании генетиче-
ской нестабильности опухолевой клетки не только за счет повышения частоты точечных мутаций. Что каса
ется механизма |
глобального демети |
лирования Д Н К |
в неопластических |
клетках, то он остается пока не ясным.
* * *
Итак, характерной чертой опухо левых и трансформированных in vitro клеток является нарушение баланса метилирования Д Н К . Дисбаланс вы ражается в том, что в одном типе опухолевых клеток наблюдаются аберрантное гиперметилирование промоторных зон генов, вовлеченных в канцерогенез, и снижение уровня метилирования остальной части ге нома.
На первый взгляд, одновременное присутствие этих двух типов наруше ний кажется парадоксальным. Для по нимания этого парадокса необходимо исследование механизмов координа ции систем метилирования и демети лирования в эмбриогенезе. Оба типа нарушений вносят свой вклад в созда ние или фенотипической, или генети ческой нестабильности опухолевой клетки. Эта особенность метилирова ния, являясь эпигенетической моди фикацией Д Н К , может в случае нару шения приводить к генетическим из менениям и поднимает вопрос о взаи модействии как генетических, так и эпигенетических процессов при воз никновении и прогрессии опухоли, показывая всю условность разделения их роли.
Рекомендуемая литература
Лихтенштейн А. В., Киселева Н. П. Мети лирование Д Н К и канцерогенез // Биохимия. — 2001. - № 66. - С. 293— 317.
Baylin S. В., Herman J. G, Graff J. R. et al.
Alterations in DNA methylation: a funda mental aspect of neoplasia // Advances
Cancer Res. - 1998. - |
Vol. |
72. - |
P. 141-198. |
|
|
Herman J. G. Hypermethylation |
of |
tumor |
202
suppressor genes in cancer // Seminar in cancer biology. — 1999. — Vol. 9. — P. 359-367.
Kass S. U., Pruss D., Wolffe A. P. How does DNA methylation repress transcription? // Trends. Genet. — 1997. — Vol. 13. - P. 444-449.
Robertson K. D., Wolffe A. P. D N A methyla
tion in health and disease. Nature re views // Genetics. — 2000. — Vol. 1,
N 11. - P. 11 |
- 19 . |
|
|
|
Rountree |
M. R., |
Bachman |
К. E., |
Herman |
J. G, |
Baylin S. B. D N A methylation, ch- |
|||
tomatin inheritace, and cancer // Onco |
||||
gene. - 2001. |
- Vol. |
20. - |
P. 3156— |
|
3165. |
|
|
|
|
Г л а в а 4
ХИМИЧЕСКИЙ КАНЦЕРОГЕНЕЗ
4 . 1 . Механизмы действия и классификация химических канцерогенов
В.С. Турусов, Г. А. Белицкий,
Л.Н. Пылев, В. А. Кобляков
Химические канцерогены, включая гормоны, ответственны за возникно вение до 80—90 % всех злокачествен ных опухолей человека. По механиз мам своего действия химические кан церогены подразделяются на генотоксичные и негенотоксичные. Незави симо от механизмов действия канце рогена для возникновения опухолей необходимы наследуемые изменения в клеточном геноме в виде специфиче ских мутаций, возникающих в случае генотоксичных канцерогенов в ре зультате прямого взаимодействия ме таболита канцерогена с ДНК, а в слу чае негенотоксичных — в результате спонтанных мутаций или вторичных влияний на ДНК (оксидативный стресс, комплексы канцероген—ре цептор и др.). Механизмы действия канцерогенов этих классов будут осве щены в данном разделе.
Канцерогенез в настоящее время большинством исследователей рас сматривается как многостадийный процесс, в котором следует различать три главные стадии: инициацию, про моцию и прогрессию. Первые две ста дии выделили еще в 1940-х годах I. Berenblum и P. Shubik, и эти термины продолжают использоваться экспери ментаторами-онкологами до настоя щего времени. Сохранили свое значе ние и некоторые постулаты гипотезы о двухстадийном канцерогенезе, а именно: 1) существует два типа аген
тов (инициаторы и промоторы), раз личающиеся по механизмам своего действия; 2) действие инициаторов необратимо, действие промоторов до определенного момента обратимо. Ос тался неизменным и порядок приме нения агентов в эксперименте (снача ла инициатор, а потом промотор), ко гда желают установить, к какому из них относится испытуемое вещество. Промоторы не способны быть ини циаторами, большинство "сильных" канцерогенов обладают и инициирую щими, и промоторными свойствами. Не подтвердился вывод о неканцерогенности промоторов: все они, за ред кими исключениями, проявляют кан церогенную активность, если их при менять в высоких дозах и достаточно долго. Деление на инициаторы и про моторы в определенной степени соот ветствует делению канцерогенов на генотоксические и негенотоксиче-
кие.
4.1.1. Генотоксичные канцерогены
Генотоксичные вещества делят на канцерогены прямого действия и со единения, неканцерогенные в исход ной форме, но активирующиеся в клетке под действием соответствую щих ферментов. Они обозначаются как непрямые канцерогены.
Канцерогены прямого действия при
растворении в жидкостях с высокой диэлектрической постоянной (в пер вую очередь в воде) распадаются с об разованием высокоактивных произ водных, содержащих в структуре из быточный положительный заряд (электрофильную группу). Например:
204

Электрофильная |
группа |
взаимо |
тельно легко удаляется из ДНК в ходе |
|||||||
действует с отрицательно |
заряженны |
репарации. |
|
|
|
|||||
ми (нуклеофильными) группами мо |
В |
нормальных |
|
клетках млекопи |
||||||
лекулы ДНК, образуя стабильную ко- |
тающих имеется фермент Ο-6-метил- |
|||||||||
валентную |
связь. |
При |
репликации |
трансфераза (Ο-6-МТаза), который |
||||||
нуклеотид, связанный с остатком кан |
удаляет Ο-6-метиладдукты из гуанина |
|||||||||
церогена, |
может быть |
неправильно |
ДНК. Различия в уровне этого фер |
|||||||
считан ДНК-полимеразой, что приво |
мента |
между видами, индивидуумами |
||||||||
дит к мутации. |
|
|
|
|
и органами у одного и того же инди |
|||||
К агентам |
такого рода относятся |
видуума огромны. У человека эти |
||||||||
Ν-нитрозоалкилмочевины |
|
(НАМ), |
уровни, как правило, выше, чем у |
|||||||
этил- |
и |
метил-метансульфонат |
грызунов. |
Характер |
различий между |
|||||
(ММС), N-метил-М-нитронитрозо- |
органами у крыс и человека в прин |
|||||||||
гуанидин |
(МННГ) |
азотистый |
иприт, |
ципе сходный: активность Ο-6-МТазы |
||||||
диэпоксибутан, |
бета-пропиолактон, |
наивысшая в печени, самая низкая в |
||||||||
этиленамин. Некоторые из них явля |
головном |
мозге |
с |
промежуточными |
||||||
ются доказанными канцерогенами че |
значениями в почках, селезенке, ки |
|||||||||
ловека. Результат действия этих со |
шечнике. Это в какой-то степени кор |
|||||||||
единений на ДНК — образование ад- |
релирует с органотропностыо нитро- |
|||||||||
дуктов, два из которых — Ν-7-метил- |
зоалкилмочевины |
при индукции опу |
||||||||
гуанин и Ο-6-метилгуанин — наибо |
холей крыс. Без дополнительных воз |
|||||||||
лее изучены. Эффективность |
индук |
действий НММ не вызывает у них |
||||||||
ции опухолей коррелирует с уровнем |
опухолей печени при различных путях |
|||||||||
содержания и длительностью перси- |
введения, тогда как частота опухолей |
|||||||||
стенции последнего аддукта, хотя пер |
мозга при тех же условиях может быть |
|||||||||
вый образуется в значительно боль |
достаточно высокой. Связь органо- |
|||||||||
шем количестве, но при этом сравни- |
тропности канцерогена с уровнем экс- |
|||||||||
С х е м а 4.1. Последовательность действия химических канцерогенов
/ фаза активации
205
прессии Ο-6-МТазы наблюдается лишь для некоторых прямых алкилирующих соединений и не распростра няется на нитрозамины, подвергаю щиеся метаболической активации.
Для каждого класса канцерогенов существуют свои пути метаболизма, конечные активные метаболиты и об разуемые ими аддукты. Последние не редко используются как маркеры воз действия канцерогенов (в молекуляр ной эпидемиологии).
Канцерогены непрямого действия.
Факт включения остатков этих малореакционноспособных соединений в макромолекулы клетки ставил в тупик исследователей до тех пор, пока в 1956 г. супруги Миллер не высказали предположение, что эти вещества в процессе метаболизма подвергаются ферментативной активации с образо ванием высокоактивных электрофильных метаболитов, способных взаимо действовать с нуклеофильными груп пами ДНК.
Последовательность стадий хими ческого канцерогенеза приведена на схеме 4.1.
4 . 1 . 1 . 1 . Метаболическая активация и детоксикация химических канцерогенов
Канцерогены непрямого действия метаболизируются в клетке специаль ными ферментными системами. Эти системы эволюционировали в ходе исторического развития в направле нии не только метаболизма эндоген ных субстратов, но и нейтрализации чужеродных соединений (ксенобиоти ков). Большинство проканцерогенов гидрофобны, поэтому способ их выве дения из клетки сводится в основном к повышению водорастворимости. При этом первым событием в цепи метаболических превращений являет ся окисление исходной молекулы. Эта реакция осуществляется в основном изоформами цитохрома Р450. Продук ты окисления подвергаются дальней шему превращению с образованием парных соединений, которые еще лег
че выводятся из клетки и организма. Этот этап осуществляется эпоксидгидролазами, глутатион-8-трансфера- зами, сульфатазами, ацетил- и глюкуронилтрансферазами и др. Наряду с основным процессом детоксикации ряд соединений в ходе этих реакций активируется, превращаясь в непо средственные канцерогены — высоко реактивные производные, ковалентно связывающиеся с клеточными белка ми и нуклеиновыми кислотами.
Как правило, электрофильные ме таболиты образуются на первом этапе окисления проканцерогена микросомными монооксигеназами, которые локализованы главным образом в эндоплазматическом ретикулуме и име ют в качестве терминального звена цитохром Р450.
Цитохром Р450, открытый в 50-е годы Клингенбергом, является инду цируемым гемопротеином, восстанов ленная форма которого при соедине нии с окисью углерода имеет в спек тре поглощения характерный пик на длине волны 450 нм.
В организме цитохром Р450 суще ствует в виде множества изоформ (в настоящее время известно более 400), обладающих различной, иногда пере крывающейся субстратной специфич ностью и различными механизмами регуляции экспрессии. Первоначаль но изоформы цитохрома Р450 класси фицировали в зависимости от их чув ствительности к индукторам и инги биторам, а после расшифровки нуклеотидной последовательности — по степени гомологии. Суперсемейство цитохрома Р450 делится на семейства, имеющие 40 % гомологии, подсемей ства с 55—60 % гомологии и отдель ные изоформы. В соответствии с при нятой номенклатурой семейства обо значают римскими цифрами, подсе мейства — заглавными буквами ла тинского алфавита, а отдельные изо формы — арабскими цифрами. Каж дая изоформа имеет свой спектр метаболизируемых субстратов. При этом один и тот же ксенобиотик может метаболизироваться различными изо-
206
формами с образованием одинаковых или разных производных. В зависимо сти от преобладания в клетке тех или иных изоформ изменяется спектр ме таболизма проканцерогена, в частно сти соотношение продуктов актива ции и детоксикации.
Следует отметить, что каждая изоформа цитохрома Р450 у индивидуу мов одного и того же вида может быть представлена набором вариантов, от личающихся по своей активности. В результате обеспечивается полимор физм, необходимый для адаптации вида к меняющимся условиям внеш ней среды. Считается, что основой возникновения такого полиморфизма является мутагенез. Наличие разли чающихся аллелей данного цитохрома в значительной степени обусловливает существование межвидовых, внутри видовых, половых и тканевых разли чий в чувствительности к канцероге нам. Функциональный цикл цитохро ма Р450 представлен на схеме 4.1. Ци тохром Р450 в этой системе работает как конечная оксидаза, катализирую щая включение одного атома кисло рода в субстрат, а второго — в обра зующуюся молекулу воды. Окислен ная форма Р450 вначале взаимодейст вует с субстратом. Затем электрон от НАДФ-Н2 через другой компонент монооксигеназной ферментной систе мы — НАДФ-цитохром-с-редуктазу — восстанавливает этот комплекс, кото рый становится способным связывать молекулярный кислород. Присоеди ненный кислород отбирает электрон от гема, и в комплексе образуется су пероксид. Этот тройной комплекс (оксицитохром) подвергается одному из двух возможных превращений:
•основной путь связан с получе нием второго электрона также от НАДФ-цитохром-с-редуктазы и восстановлением гема с после дующим стереоспецифическим окислением субстрата;
•оксицитохром распадается (ре акция "разобщения") до получе ния второго электрона с высво
бождением исходного ферментсубстратного комплекса и ак тивных форм кислорода — су пероксида, перекиси водорода, гидроксилрадикала.
Образование активных форм ки слорода в клетке также является од ним из факторов канцерогенеза.
Окисленный на цитохроме Р450 субстрат подвергается воздействию ферментов конъюгации, катализирую щих включение в структуры вещества глутатиона, серной кислоты, глукуроновой кислоты и пр. В результате об разованный метаболит становится гидрофильным и легко выводится из организма с мочой. При этом, однако, на некоторых этапах превращения об разуется высокоактивное вещество, способное взаимодействовать с мак ромолекулами, оказывая канцероген ный или токсический эффект.
Основные изоформы Р450, метаболизирующие канцерогены у человека, представлены в табл. 4.1. С опреде ленной долей приближения их актив ность может быть измерена in vivo по уровню метаболизма соответствующих неканцерогенных субстратов.
Цитохром CYP1A1 является клю чевым ферментом в метаболизме кан церогенных полициклических арома тических углеводородов, которые яв ляются его индукторами. У человека он обнаруживается в значимом коли честве только в легких курильщиков. Цитохром CYP1A2 экспрессируется в ткани печени, где его индуцируют те же ксенобиотики, что и CYP1A1. По мимо лекарственных препаратов типа фенацетина, ацетаминофена и кофеи на, он необходим для метаболической активации проканцерогенных ариламинов и гетероциклических аминов, образующихся при термической обра ботке пищи. CYP2A6 и CYP2B6 экспрессируются в печени и других орга нах. Они могут активировать циклофосфамид и канцерогенные афлатоксины. CYP2A6 активирует также не которые канцерогенные нитрозамины и в их числе табакоспецифический
207
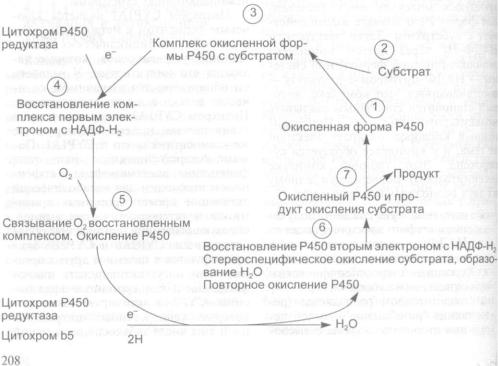
Т а б л и ц а 4.1. Основные изоформы цитохрома Р450, |
метаболизирующие канцерогены |
|||||||
у человека |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
, Изо- |
|
|
Субстраты |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
форма |
|
канцерогенные |
|
|
|
|
неканцерогенные |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
||
CYP1A1 |
Канцерогенные ПАУ |
|
|
|
|
Неканцерогенные ПАУ |
||
CYP1A2 |
Афлатоксины, гетероциклические амины |
|
|
|
Кофеин |
|
||
CYP2A6 |
Циклофосфамид, нитрозамины, афлатоксины |
|
|
Кумарин и его производные |
||||
CYP2B6 |
|
|
|
|
|
|
|
|
CYP2D6 |
Нитрозамины, в том числе табакоспецифические |
|
Дебризоквин, 5-мефенитон |
|||||
CYP2E1 |
Низкомолекулярные соединения, в том числе |
|
|
Галогенизированные ане |
||||
|
бензол, винилхлорид, нитрозамины |
|
|
|
стетики, этанол |
|||
CYP3A4 |
Афлатоксины, дигидродиолы нитрополиаренов, |
|
Нефедипин, эстрогены |
|||||
|
гетероциклические амины |
|
|
|
|
|
|
|
|
|
|
|
|||||
4-(метилнитрозамино)-1 -(3-пиридил)- |
ности изоформа CYP3A4, метаболизи- |
|||||||
1-бутанон. Индуцируемый этанолом |
рует |
проканцерогенные |
афлатокси |
|||||
CYP2E1 |
активирует |
многие канцеро |
ны, |
а также |
превращает |
многие ди |
||
генные |
соединения |
низкомолекуляр |
гидродиолы |
полициклйческих арома |
||||
ного строения, такие как бензол и ви |
тических |
углеводородов |
в активные |
|||||
нилхлорид, а также некоторые нитро |
диолэпоксиды (схема 4.2). |
|||||||
замины. Подсемейство CYP3A, в част |
Реакции второго этапа метаболиз- |
|||||||
С х е м а |
4.2. Окисление субстратов в системе цитохрома |
Р450 [Goldstein, Faletto, |
||||||
1993] |
|
|
|
|
|
|
|
|
ма ксенобиотиков, как уже упомина лось, сводятся в основном к образова нию конъюгатов, легко выводимых из организма.
Ν-гидроксилирование ариламинов и ацетиламинов, таких как бензидин, 2-ацетиламинофлуорен и гетероцик лические амины, образующиеся при термической обработке пищи, являет ся первым необходимым этапом их метаболического превращения в непо средственные канцерогенные ради калы.
S-окисление, деалкилирование,
эпоксидирование олефинов, изменяю щие биологические свойства ряда ши роко распространенных соединений, также входят в круг реакций, катали зируемых изоформами цитохрома Р450. Реакции второго этапа метабо лизма ксенобиотиков, как уже упоми налось, сводятся в основном к образо ванию конъюгатов, легко выводимых из организма.
Эпоксидгидролазы осуществляют промежуточный этап детоксикации образованных цитохромом Р450 эпоксидов, которые превращаются в трансдигидродиолы и далее с помо щью других ферментов в конъюгаты с глюкуроновой кислотой, глутатионом или сульфатом. Некоторые соедине ния при этом, наоборот, превращают ся в активные канцерогенные метабо литы. Гидрирование 7,8-оксида бенз(а)пирена до дигидродиола с дальнейшим образованием из него с помощью цитохрома Р450 диолэпоксида приводит к активации молекулы и ее превращению в непосредствен ный канцерогенный метаболит. Суще ственно, что индивидуальные показа тели активности этих ферментов у че ловека могут различаться в десятки раз.
Глутатион-8-трансферазы катали зируют перенос глутатиона на электрофильные соединения. Это приво дит к нейтрализации электрофилов, образующихся из бензола, афлатоксина, ПАУ, и к активации дибромэтилена или хлористого метилена.
УДФ-глюкуронилтрансферазы.
Ферменты этой группы также инактивируют многие электрофильные со единения, но активируют производ ные гетероциклических аминов, обра зующихся в пище при ее жарении. Считается, что эти соединения играют существенную роль в возникновении рака толстого кишечника у человека. Они вначале окисляются по азоту, за тем превращаются в Ν-глюкурониды. В кишечнике комплексы расщепля ются р-глюкуронидазой и в конечном итоге превращаются в высокореактив ные электрофилы — Ν-ацетоксиари- ламины.
Ацетилтрансферазы ацетилируют аминогруппы ариламинов и гидрази нов, что является необходимым эта пом их активации и/или детоксика ции. При этом 2-аминофлуорен акти вируется, а 2-нафтиламин и бензидин детоксицируются. Изоформы этого фермента, преобладающие у отдель ных индивидуумов, отличаются по ак тивности в десятки раз, поэтому в че ловеческой популяции различают ак тивные и неактивные (или быстрые и медленные) ацетиляторы. У первых профессиональный рак мочевого пу зыря, индуцируемый 2-нафтиламином и бензидином, развивается значитель но реже, чем у вторых.
Образование сульфатов также от носится к реакциям детоксикации. Обратный эффект наблюдается реже, например при образовании Ν-сульфо- нилоксипроизводных 2-ацетилами- нофлуорена.
4.1.1.2. Полициклические ароматические углеводороды
К этому классу химических ве ществ относятся соединения с кон денсированными бензольными ядра ми. Степень канцерогенности зависит от числа ароматических колец в моле куле, взаимного их расположения, на личия заместителей в молекуле.
Наибольшей канцерогенностью обладают вещества, имеющие 4—7 бензольных конденсированных колец. В структуре полициклических арома-
14-7908 Д. Г. Заридзе |
209 |
С х е ма 4.3. Примеры структур ароматических углеводородов
|
|
|
|
|
|
|
Антантрен |
|
|
|
|
|
|
|||
тических углеводородов |
выделены |
зо |
тронном восстановлении, или карбо- |
|||||||||||||
ны, придающие соединению канцеро |
ний-катион, образование которого |
|||||||||||||||
генную активность, — так называемые |
обусловлено |
трехстадийным |
процес |
|||||||||||||
бей- и фьорд-области (схема 4.3), од |
сом: |
|
|
|
|
|
||||||||||
нако |
имеются |
вещества, |
обладающие |
• |
ферментативное |
метилирование |
||||||||||
канцерогенностью (например, |
антан |
|||||||||||||||
|
вещества; |
|
|
|||||||||||||
трен), |
в |
структуре |
которых |
отсутству |
|
|
|
|||||||||
• |
окисление метальной группы; |
|||||||||||||||
ют бей- и фьорд-области. |
|
|
|
|||||||||||||
|
|
|
• |
образование сульфоэфира с по |
||||||||||||
Введение метильной |
группы, |
как |
||||||||||||||
|
следующим спонтанным отщеп |
|||||||||||||||
правило, |
увеличивает |
|
канцероген- |
|
||||||||||||
|
|
лением сульфогруппы и образо |
||||||||||||||
ность |
соединения, |
а |
|
галогенов |
— |
|
||||||||||
|
|
ванием карбоний-катиона. |
||||||||||||||
уменьшает. Наличие метильной груп |
|
|||||||||||||||
По-видимому, возможностью об |
||||||||||||||||
пы приводит к усилению канцероген |
||||||||||||||||
ности только в тех случаях, когда по |
разования |
различных типов активных |
||||||||||||||
ложение, в которое она введена, не |
метаболитов из одного и того же со |
|||||||||||||||
препятствует |
активации |
соединения |
единения объясняется низкая органо- |
|||||||||||||
до конечного канцерогена. Так, на |
и видоспецифичность этого класса ве |
|||||||||||||||
пример, введение метильной группы в |
ществ, а также канцерогенность ве |
|||||||||||||||
положение 7 или 12 молекулы |
ществ, не имеющих в структуре бей- и |
|||||||||||||||
бенз(а)антрацена усиливает его канце |
фьорд-области. |
|
|
|||||||||||||
рогенный потенциал, а введение в по |
Нитроарсны имеют в структуре по |
|||||||||||||||
ложение 7 или 8 молекулы бенз(а)пи- |
лициклическое ароматическое ядро и |
|||||||||||||||
рена |
ослабляет активность |
послед |
нитрогруппу. Несмотря на то что ПАУ |
|||||||||||||
него. |
|
|
|
|
|
|
|
|
|
являются |
сильными |
канцерогенами, |
||||
Наиболее широкую поддержку по |
большинство |
исследователей |
считают, |
|||||||||||||
лучило представление, |
согласно кото |
что эта составляющая молекулы нит- |
||||||||||||||
рому конечным канцерогенным мета |
роаренов не участвует в образовании |
|||||||||||||||
болитом полициклических ароматиче |
конечного |
канцерогена. Окисление |
||||||||||||||
ских углеводородов является диолэ- |
ароматической части молекулы нит- |
|||||||||||||||
поксид, образованный по бей-облас |
роаренов |
является процессом деток |
||||||||||||||
ти. Им может быть также радикал-ка |
сикации, а не активации. Образование |
|||||||||||||||
тион, |
образующийся |
при |
одноэлек- |
конечного канцерогена связано с вос- |
||||||||||||
210

становлением нитрогруппы в амино группу, а далее это аминосоединение подвергается ферментативным воз действиям, идентичным тем, которые происходят с ароматическими ами нами.
Как загрязнители атмосферы нитроарены выявлены сравнительно не давно. Они определяются в отрабо танных газах бензиновых и дизель ных двигателей. Их канцерогенный потенциал довольно велик: они обла дают широким спектром действия, вызывая у экспериментальных жи вотных опухоли как на месте введе ния, так и в отдаленных органах. Канцерогенная опасность нитроаренов для человека не установлена вследствие отсутствия эпидемиологи ческих данных, однако содержание их в заведомо канцерогенных смесях, какими являются выбросы двигате лей, делает эти соединения подозри тельными в отношении канцероген ной опасности для человека. При оп ределении нитроаренов в смесях в качестве маркерного вещества ис пользуют 6-нитрохризен.
4.1.1.3. Нитрозосоединения
Magee и Barnes в 1956 г. впервые продемонстрировали канцерогенность нитрозосоединений на печени крыс: они вызывают опухоли у всех видов животных, на которых испытывались. Известно более 300 канцеро генных нитрозосоединений, которые широко распространены в окружаю щей среде (пища, напитки, космети ческие средства, табачный дым, атмо сферный воздух на некоторых произ водствах и вокруг них и т. д.). Могут синтезироваться в желудке из алкиламинов и нитратов. Их способность вызывать злокачественные опухоли у человека доказана более или менее твердо лишь в отношении табачных нитрозосоединений.
По механизмам своего действия они разделяются на нитрозамины, ак тивируемые системой микросомальных оксидаз, и нитрозамиды, подвер
14*
гающиеся спонтанному распаду также с образованием алкилирующего иона.
Нитрозамины. Начальным этапом метаболической активации НДМА яв ляется образование нестойкого а-гид- роксинитрозамина. Его разложение ведет к образованию метилнитрозамина, затем формальдегида и сильного алкилирующего агента — метилдиазогидроксида, который диссоциируется на азот и метилкарбониевый ион, ме тилирующий белки и ДНК. В этот же конечный продукт превращается и 1,2-диметилгидразин — канцероген, близкий нитрозаминам.
Нитрозамиды. Как говорилось вы ше, нитрозамиды подвергаются в ор ганизме спонтанному распаду с обра зованием продуктов, метилирующих ДНК. Между некоторыми из них (на пример, между НММ и ММС) име ются существенные различия в харак тере их реакций с атомами кислорода. НММ относят к соединениям, обра зующим карбониевый ион и реаги рующим с кислородными нуклеофильными центрами в ДНК. Напро тив, ММС относится преимуществен но к соединениям, не образующим карбониевого иона и реагирующим с "мягкими" азотными нуклеофильными центрами. Эти особенности отра жаются и на канцерогенной активно сти указанных соединений: однократ ная доза НММ вызывает у мышей вы сокую частоту тимусных лимфом, то гда как эквимолярная доза ММС во обще не канцерогенна, хотя общий объем алкилирования в обоих случаях одинаков. Анализ тимусной ДНК по казал прямую корреляцию между сте пенью образования Ο-6-метилгуанина (высокая в случае НММ) и частотой опухолей.
4.1.1.4. Ароматические амины
Канцерогенность ароматических аминов (бывших причиной рака моче вого пузыря у рабочих, занятых в про изводстве красителей) была впервые установлена в конце прошлого века в Германии. Известны три основных
211
ароматических амина, вызывающих рак мочевого пузыря у человека: 2- нафтиламин, бензидин и 4-аминоби- фенил. Последний обнаруживают, хо тя и в ничтожном количестве, в табач ном дыме, с чем, по-видимому, свя зан повышенный риск рака мочевого пузыря у курильщиков (см. раздел 4.2). В крови человека постоянно об наруживают гемоглобиновые аддукты 4-аминобифенила, что свидетельству ет об экспозиции человека ароматиче ским аминам, источник которой неиз вестен.
Метаболическая активация арома тических аминов начинается с их ацетилирования с помощью цитозольного АсСоА до ароматических амидов. Реакция может быть обрати мой с деацетилированием амидов до ароматических аминов. Затем проис ходит микросомальное Ν-окисление ароматических аминов до Ν-гидро- ксиариламинов (они могут напрямую взаимодействовать с ДНК) и окисле ние ароматических амидов до арилгидроксаминовых кислот. Послед ние подвергаются дополнительной метаболической активации, которая заключается в структурной пере стройке до Ν-ацетоксиариламина с помощью ацилтрансферазы или до Ν-сульфонилоксиариламина с помо щью сульфотрансферазы. Конечны ми продуктами являются ацетилированные в случае амидов и неацетилированные в случае аминов аддукты ДНК, образующиеся при ковалентном присоединении азота к С8-де- зоксигуанозину.
Наиболее обстоятельно из всех ароматических аминов исследован 2-ААФ, Показано, что Ν-гидрокси- лирование является начальным про цессом активации 2-ААФ с образова нием Ν-гидрокси-ААФ. Этот метабо лит характерен для мышей, крыс и хомячков, чувствительных к канцеро генному действию 2-ААФ. У морских свинок, резистентных к 2-ААФ, от сутствует микросомальный фермент, активирующий 2-ААФ до Ν-гидро кси-ААФ.
4.1.1.5. Афлатоксины
Эти канцерогены являются про дуктом гриба Aspergillus flavus, расту- ] щего на земляных орехах, злаках. Су ществует несколько природных афлатоксинов, из которых наиболее мута- ] генным и канцерогенным, а также лучше всех изученным является афлатоксин В, (AFB,). Именно с AFB,, часто в сочетании с вирусом гепатита В, связывают высокую частоту рака печени в Африке и Азии.
Как и рассмотренные выше канце рогены, AFB, нуждается в метаболи ческой активации, проходящей ряд стадий с образованием нескольких метаболитов (в том числе ΑΡΒ,-диола) и нескольких аддуктов ДНК.
4.7.2. Негенотоксические канцерогены
Речь идет о большой группе хими ческих соединений, способных вызы вать злокачественные опухоли у лабо раторных животных, но обладающих слабой генотоксичностью или не об ладающих ею в тестах, в которых они испытывались. Противоречивость словосочетания "негенотоксический канцероген" очевидна: если вещество индуцирует опухоли, оно, следова тельно, реагирует с ДНК, вызывая специфическую мутацию; если же оно не вызывает мутации, то не должно быть канцерогеном. Несмотря на ус ловность этих терминов (негеноток сический, или эпигенетический), они прочно вошли в обиход, и под ними обычно понимают не полное отсутст- вие генотоксичности, а неспособность соединения ковалентно связываться с ДНК и образовывать аддукты. Например, каждый из трех негенотоксических канцерогенов (фенобарбитал, полихлорбифенилы, хлордан) вызвал положительные реакции в отдельных тестах на генотоксичность in vitro, но все три оказались неспособными об разовывать аддукты ДНК. В противо положность этому генотоксические канцерогены, особенно не нуждаю-
212
щиеся в метаболической активации, дают положительные результаты в большинстве или даже во всех тестах на генотоксичность in vivo и in vitro, в которых они исследовались.
К негенотоксическим канцероге нам относятся соединения различной химической структуры и различного механизма действия: промоторы двухстадийного канцерогенеза, СПП, пес тициды, гормоны, волокнистые мате риалы, прочие соединения (нужно за метить, что и пестициды, и гормоны могут быть промоторами канцероге неза).
Негенотоксические канцерогены часто называют канцерогенами промоторного типа. Промоторы, как уже говорилось, в эксперименте должны применяться в высоких дозах, дли тельно и, что очень важно, беспре рывно. Более или менее длительный перерыв в их применении сопровож дается остановкой канцерогенеза (но вые опухоли больше не появляются) или даже регрессией возникших опу холей. Они вызывают клеточную про лиферацию, тормозят апоптоз, нару шают взаимодействие между клетками.
Промоторы канцерогенеза многих органов сами по себе вызывают опу холи и без предварительного действия инициатора: кретоновое масло и вы деленный из него ТФА вызывают опу холи кожи, фенобарбитал — опухоли печени, сахарин — опухоли мочевого пузыря, гойтрогенные агенты (аминотриазол, метилтиоурацил, дефицит йода и др.) — опухоли щитовидной железы. Канцерогенность промото ров обычно принято объяснять про моцией спонтанной инициации. Предлагается, однако, и другое объяс нение, а именно: сам промотор может обладать не только промоторной, но и слабой инициирующей активностью, которую можно приписать положи тельной реакции многих промоторов в одном, а то и в нескольких тестах на генотоксичность. Другие авторы ут верждают, что канцерогенность негенотоксичных агентов всегда определя ется негенотоксическим механизмом
даже при наличии у них признаков генотоксичности. Так или иначе, все промоторы, проявившие канцероген ную активность, могут быть отнесены к эпигенетическим канцерогенам, но не все эпигенетические канцерогены испытывались на промоторную актив ность.
4 . 1 . 2 . 1 . Особенности канцерогенного эффекта негенотоксичных канцерогенов
Негенотоксичные канцерогены вы зывают у животных развитие доброка чественных и злокачественных опухо лей, однако по сравнению с генотоксичными канцерогенами для достиже ния канцерогенного эффекта требу ются определенные условия.
1. Неэффективность однократного воздействия. Многие генотоксические канцерогены (ДМБА, нитрозоалкилмочевины, нитрозамины и др.) спо собны вызывать опухоли при одно кратном воздействии или быть ини циаторами двухстадийного канцероге неза. Подобными свойствами не обла дают негенотоксические (эпигенети ческие) канцерогены.
2.Необходимость больших доз й длительного беспрерывного воздейст вия — особенность практически всех или по крайней мере большинства эпигенетических канцерогенов. Прав да, ДДТ вызывает у мышей высокую частоту опухолей печени уже при дозе 2 мг/кг массы тела, что, вероятно, связано с его стабильностью и спо собностью накапливаться в тканях. В целом же различия в эффективных дозах между генотоксическими и не-
генотоксическими канцерогенами в крайних случаях достигают 106.
3.Остановка канцерогенеза при прекращении воздействия. В случае генотоксических канцерогенов пре кращение воздействия канцерогена ведет к замедлению появления новых опухолей, но они продолжают появ ляться, хотя и после весьма длитель ного латентного периода. Пример — развитие так называемого профессио-
213
нального рака через десятилетия по сле прекращения контакта с произ водственным канцерогеном.
При прекращении действия негенотоксических канцерогенов происходит не только замедление канцерогенеза, но и его полная остановка. В США длительное время применяли эстроге ны в качестве заместительной терапии
вклимактерическом периоде, что при вело к повышению частоты рака эндо метрия. Это повышение было отмечено
в1975 г., после чего употребление эст рогенов начало быстро снижаться, за чем последовало быстрое снижение риска рака эндометрия.
Вэксперименте применение ДДТ в течение ограниченного отрезка време ни (40 нед) привело к повышению частоты опухолей печени, которая ос тавалась на том же уровне при наблю дении за животными в течение после дующих полутора лет.
Характер изменения риска рака легкого у бросивших курить иллюст рирует оба варианта: в течение 5 лет после прекращения курения происхо дит значительное сокращение риска рака за счет, вероятно, промоторов и эпигенетических канцерогенов табач ного дыма. В последующем, однако, риск начинает возрастать (это эффект генотоксических канцерогенов, содер жащихся в табачном дыме), сохраня ясь на более высоком уровне по срав нению с теми, кто никогда не курил.
В80-е годы стали различать канце рогены ранних и поздних стадий. Прекращение действия первых ведет лишь к некоторому замедлению кан церогенеза, тогда как прекращение действия вторых дает быстрый эффект
ввиде остановки канцерогенеза. В зна чительной мере это совпадает с разли чиями в действии генотоксических и негенотоксических канцерогенов.
4.1.2.2. Механизмы действия негенотоксичных канцерогенов
Известны следующие механизмы действия негенотоксичных канцероге нов:
а) промоция спонтанной инициа ции;
б) цитотоксичность со стойкой клеточной пролиферацией (митогенный эффект);
в) оксидативный стресс; г) образование комплекса канце
роген—рецептор; д) торможение апоптоза;
ж) нарушение межклеточных ще левых контактов.
Следует заметить, что в канцеро генном действии одного и того же со единения могут принимать участие несколько механизмов.
Промоция спонтанной инициации —
по-видимому, наиболее распростра ненный механизм канцерогенеза сре ди негенотоксических канцерогенов.
Цитотоксичность. Многие эпигене тические канцерогены обладают цитотоксическими свойствами. У некото рых из них, например у фурана и хло роформа, они выражены настолько резко, что им приписывают роль в развитии опухолей. При канцерогене зе, индуцированном хлороформом, опухоли печени и почек развивались лишь при действии больших разовых доз, вызывавших некрозы и компен саторную клеточную пролиферацию, и не развивались при введении хлоро форма с питьевой водой, т. е. в малых разовых дозах, когда цитотоксичность отсутствовала. Предполагают, что массовая гибель клеток, вызванная цитотоксичностью, ведет к пролифе рации выживших клеток, в том числе мутировавших, с конечным развитием опухоли.
Оксидативный стресс с образова нием перекиси водорода и молекул активного кислорода — механизм дей ствия многих негенотоксических кан церогенов, в частности СПП, волок нистых канцерогенов.
Образование комплекса канцеро
ген—рецептор. Подробно об этом ме ханизме изложено в разделе о стиму ляторах пролиферации пероксисом (см. раздел 3.2.3.).
Торможение апоптоза — признан-
214
ный ныне эффект действия негено токсичных канцерогенов, например фенобарбитала, диоксина, СПП.
Нарушение межклеточных щелевых контактов. Эти нарушения ведут к изо ляции опухолевой клетки от нормаль ных окружающих клеток и их контро лирующего пролиферацию влияния.
Генотоксический и негенотоксический механизмы постоянно взаимодей ствуют в процессе канцерогенеза, и один и тот же эффект может быть вы зван посредством как генотоксического, так и негенотоксического механиз ма. Самый убедительный пример взаи модействия — двухстадийный канцеро генез кожи: генотоксичный инициатор вызывает мутацию протоонкогена, то гда как промотор действует через негенотоксический механизм, влияя на межклеточные взаимодействия, вызы вая образование молекул активного ки слорода и т. д. Более того, каждый без исключения генотоксический канцеро ген обладает не только инициирую щей, но и промоторной (негенотоксической) активностью.
4.1.2.3. Классификация негенотоксических канцерогенов по механизму их действия
В отличие от генотоксических кан церогенов, обладающих единым ко нечным механизмом, группы негено токсических канцерогенов и их от дельные представители могут обладать специфическими механизмами канце рогенного действия. Далее рассмотре ны следующие группы соединений:
•гормоны;
•соединения с эстрогеноподобным действием (ксеногормоны);
•тиреотропные канцерогены;
•стимуляторы пролиферации пероксисом (СПП);
•цитотоксичные соединения, вы зывающие длительную клеточ ную пролиферацию;
•ингибиторы или стимуляторы ферментных систем, участвую
щих в регуляции передачи митотического сигнала;
•соединения, стимулирующие образование активных форм ки слорода;
•прочие негенотоксичные соеди нения.
4.1.2.3.1. Гормоны
Речь идет об эстрогенах — природ ных или стильбеновых (диэтилстильбэстрол). Действие их осуществляется через специфические рецепторы. В обоих случаях в механизмах канцеро генного действия присутствует два компонента — промоторный и гено токсичный, но представленных, одна ко, в разной степени. Промоторный, или физиологический, компонент сводится к гормональной стимуляции, т. е. к вызываемой гормоном клеточ ной пролиферации в органе-мишени благодаря наличию в клетках рецепто ров эстрогенов. Но и в канцерогенном действии природных эстрогенов в ка кой-то степени присутствует геноток сичный компонент: при их метаболиз ме образуются так называемые катехолэстрогены, а из них — генотоксические метаболиты, хотя и в незна чительном количестве. При действии диэтилстильбэстрола (ДЭС) геноток сический компонент выражен весьма сильно — при его метаболизме обра зуются хиноны и семихиноны, обла дающие отчетливыми генотоксическими свойствами (подробнее см. гла ву б, "Гормональный канцерогенез"). ДЭС — канцероген человека, вызывав ший у молодых женщин, на которых он действовал in utero (его в свое время применяли для сохранения беременно сти), светлоклеточный рак влагалища.
4.1.2.3.2. Соединения с эстрогеноподобным действием (ксеногормоны)
В последнее десятилетие стали на капливаться сведения о репродуктив ных аномалиях у диких животных (пониженная плодовитость, демаску-
215
линизация, феминизация, изменения |
рые |
хлорорганические |
пестициды |
|||||||||||||
половых органов). Эти изменения |
(хлордан, |
гексахлорциклогексан, от |
||||||||||||||
приписывают |
ксенобиотикам |
окру |
дельные метаболиты ДДТ), такие пес |
|||||||||||||
жающей среды, мимикрирующим эф |
тициды, как дельтаметрин, трифлюра- |
|||||||||||||||
фекты эндогенных гормонов (эстроге |
лин и др. |
|
|
|
|
|
|
|||||||||
нов/антиэстрогенов, |
андрогенов/ан- |
Первичным эффектом этих соеди |
||||||||||||||
тиандрогенов |
и т. д.). Главным |
эф |
нений является активация ферментов, |
|||||||||||||
фектом этих соединений является на |
снижающих в крови уровень гормона |
|||||||||||||||
рушение |
гормонального |
статуса |
орга |
щитовидной железы, что влечет за со |
||||||||||||
низма, почему все они были названы |
бой усиленную продукцию тиреотроп- |
|||||||||||||||
endocrine disruptоrs. К ним относятся |
ного гормона гипофиза, который и |
|||||||||||||||
хлорорганические пестициды (в |
пер |
является индуктором |
опухолей щито |
|||||||||||||
вую очередь ДДТ, имеющий структур |
видной |
железы. |
Видовой |
особенно |
||||||||||||
ное сходство с диэтилстильбэстро- |
стью крыс является низкий уровень |
|||||||||||||||
лом), |
полихлорированные бифенилы, |
или отсутствие в крови белка, связы |
||||||||||||||
диоксины, алкилфенолы, в какой-то |
вающего |
гормон |
щитовидной железы |
|||||||||||||
степени |
пиретроиды. Имеются |
сооб |
(thyroxin binding globulin — TBG) и |
|||||||||||||
щения о более высоком содержании |
тем самым препятствующего его вы |
|||||||||||||||
метаболитов ДДТ в жировой ткани |
ведению из крови, вследствие чего не |
|||||||||||||||
молочных желез женщин, страдающих |
происходит |
стимуляции |
продукции |
|||||||||||||
раком молочной железы, по сравне |
тиреотропного гормона гипофиза. У |
|||||||||||||||
нию |
с |
контрольными |
женщинами. |
человека TBG присутствует, и, таким |
||||||||||||
Точно так же у женщин с высоким со |
образом, указанный механизм индук |
|||||||||||||||
держанием ДДЭ (метаболита ДДТ) в |
ции |
"опухолей |
щитовидной |
железы, |
||||||||||||
сыворотке крови обнаруживают |
более |
вероятнее всего, не работает. Косвен |
||||||||||||||
высокий риск заболевания раком мо |
ным |
подтверждением |
этого |
является |
||||||||||||
лочной железы по сравнению с жен |
тот факт, что эпидемиологические ис |
|||||||||||||||
щинами, у которых содержание ДДЭ |
следования |
не |
выявили |
учащения |
||||||||||||
было ниже. |
|
|
|
|
опухолей щитовидной железы у боль |
|||||||||||
Эстрогенный эффект |
ДДТ проде |
ных эпилепсией, получавших с тера |
||||||||||||||
монстрирован на лабораторных грызу |
певтической |
целью |
фенобарбитал в |
|||||||||||||
нах: отмечены |
утеротропный эффект |
дозах, сравнимых с теми, которые вы |
||||||||||||||
(крысы и мыши), развитие псевдоэст- |
зывают рак щитовидной железы у |
|||||||||||||||
руса |
(мыши), |
усиление |
эксперимен |
крыс. |
|
|
|
|
|
|
|
|||||
тального канцерогенеза молочной же |
|
|
|
|
|
|
|
|
|
|||||||
лезы, |
торможение связывания |
[3Н]- |
4.1.2.3.4. Стимуляторы |
|
|
|||||||||||
эстрадиола с |
рецептором эстрогенов |
|
|
|||||||||||||
пролиферации пероксисом (СПП) |
||||||||||||||||
in vitro (крысы). К ним же следует от |
||||||||||||||||
|
|
|
|
|
|
|
|
|
||||||||
нести и некоторые другие пестициды, |
Отличительной чертой этих соеди |
|||||||||||||||
например хлортриазины (атразин, си- |
нений (в настоящее время их насчи |
|||||||||||||||
мазин), |
не обладающие |
эстрогенной |
тывают более 71) является способ |
|||||||||||||
активностью, но нарушающие эст- |
ность |
стимулировать |
пролиферацию |
|||||||||||||
ральный цикл, вызывая персистирую- |
пероксисом в печени мышей и крыс и |
|||||||||||||||
щий эструс и тормозя продукцию ги |
одновременно вызывать у них разви |
|||||||||||||||
пофизом лютеинизирующего гормона. |
тие опухолей печени (у других видов |
|||||||||||||||
|
|
|
|
|
|
|
этот феномен не описан). В норме пе- |
|||||||||
4.1.2.3.3. Тиреотропные |
|
роксисома, принимающая, как счита |
||||||||||||||
|
ют, |
участие |
в |
метаболизме |
жирных |
|||||||||||
канцерогены |
|
|
|
|||||||||||||
|
|
|
кислот, занимает 2 % объема цито |
|||||||||||||
|
|
|
|
|
|
|
||||||||||
Целый ряд негенотоксических кан |
плазмы, под действием СПП эта циф |
|||||||||||||||
церогенов способны |
индуцировать у |
ра увеличивается до 20. В группу СПП |
||||||||||||||
крыс опухоли щитовидной железы. К |
входят |
пластификаторы, |
некоторые |
|||||||||||||
ним относятся фенобарбитал, некото |
пестициды, |
растворители |
|
(трихлор- |
||||||||||||
216
этилен), гиполипидемические средст ва (клофибрат, гемифиброзил и др.). Последние разрешены в ряде стран и применяются для похудания по 1 г/сут в течение нескольких месяцев. Опухо ли печени или других органов у таких больных не описаны. Механизмы дей ствия СПП реализуются путем обра зования активных форм кислорода (путем индукции изоформ цитохрома Р450 и/или стимуляции окисления жирных кислот в пероксисомах) или активации группы специфических ре цепторов — peroxisome proliferator-ac- tivated receptor (PPAR). В тестах на ге нотоксичность СПП дают отрицатель ные или сомнительные (лишь изредка положительные) результаты.
4.1.2.3.5. Цитотоксичные соединения, вызывающие длительную клеточную пролиферацию
Многие негенотоксические канце рогены обладают цитотоксическим действием, однако у некоторых из них это действие и связанная с ним про лиферация выражены столь сильно, что позволяют говорить о причастно сти последней к механизмам канцеро генеза. Примерами соединений, отно сящихся к этой группе, могут служить хлороформ, фуран, бутилированный гидроксианизол, пестициды хлороталонил и р-дихлорбензол. В случае хлороформа опухоли печени и почек развивались лишь при действии боль ших разовых доз, вызывавших некро зы и следовавшую за ними интенсив ную и стойкую клеточную пролифера цию. Опухоли не развивались при введении хлороформа с питьевой во дой, т. е. в малых разовых дозах, когда цитотоксичность и выраженная про лиферация отсутствовали. При дейст вии фурана развитие опухолей печени у мышей и крыс сочеталось с некроза ми, воспалением и появлением в пе чени большого количества гепатоцитов с ядрами в S-фазе.
Фунгицид хлороталонил вызыва ет опухоли преджелудка и почек: по
явлению опухолей в преджелудке предшествуют эрозии, изъязвления и длительная гиперплазия слизистой оболочки, а в почке — длительная гиперплазия проксимальных каналь цев. Во всех указанных случаях авто ры отмечали положительную корре ляцию между развитием опухолей и стойкой клеточной пролиферацией. Следует отметить, что стойкая про лиферация — характерный признак действия промоторов [Slaga и др., 1989].
4.1.2.3.6. Ингибиторы или стимуляторы ферментных
систем, участвующих в регуляции передачи митотического сигнала
Окадаиковая кислота ингибирует функцию фосфатаз — ферментов, де зактивирующих белки сигналпередающих систем клетки; в результате она вынуждена постоянно находиться в стадии деления.
Трансигаргин нарушает в клетке обмен кальция, ионы которого явля ются участниками пролиферативного каскада. Нарушения обмена кальция также ведут к нерегулируемому росту клеток, хотя исходный сигнал к деле нию может уже отсутствовать.
4.1.2.3.7. Соединения, стимулирующие образование активных форм кислорода
Образование в клетке активных форм кислорода вызывает поврежде ние ДНК, которое выражается в появ лении в структуре этой макромолеку лы окисленной формы гуанина — 8- гидроксигуанина, что является марке ром образования в клетке активных форм кислорода, однако указанное повреждение ДНК не всегда ведет к образованию опухоли.
Предполагается, что в образовании активных форм кислорода, прини мающих участие в развитии опухолей при действии многих негенотоксич ных канцерогенов, могут быть задей ствованы различные соединения:
217
•вещества, вызывающие воспале ние и стимулирующие кисло родный "взрыв" (оксидативный стресс) у макрофагов. По этому механизму принято считать, что опухоли вызывают форболовые эфиры и минеральные волокна;
•индукторы изоформ цитохрома Р450, стимулирующие образова ние активных форм кислорода этим ферментом. Наиболее вы сокий уровень индуцибельных ферментов цитохрома Р450 на ходится в печени животных и че ловека. Предполагается, что по этому механизму вызывают опу холи печени в эксперименте хлорированные бифенилы, ди оксины и фенобарбитал;
•соединения, увеличивающие ко личество митохондрий в клетке. По такому механизму, видимо, вызывает опухоли метилпирро-
видов животных) сахарин по класси фикации Международного агентства по изучению рака (МАИР) отнесен к группе 2В (возможный канцероген для человека). Считается, что человек потребляет сахарина не более 5 мг/кг в сутки, т. е. нет оснований ожидать повышения риска развития рака мо чевого пузыря у лиц, регулярно по требляющих сахарин.
d-Лимонен содержится в цитрусо вых маслах и фруктовых соках, его потребление в США составляет до 2 мг/кг в сутки. Особенностью его кан церогенного действия является индук ция аденом и рака почек только у крыс-самцов, у которых в эпителии почечных канальцев образуются гиа линовые капли, содержащие специ фический глобулин, отсутствующий у крыс-самок и других видов животных, включая человека.
4.7.3. Соединения
•соединения, стимулирующие с неустановленным пролиферацию пероксисом (о механизмом действия них подробнее см. выше);лидин;
• |
органические перекиси; которые |
4.1.3.1. Металлы |
|
|||||||
|
в клетке разлагаются с образова |
|
||||||||
|
Свойствами вызывать опухоли у |
|||||||||
|
нием активных форм кислорода. |
|||||||||
|
В эксперименте показано канце |
человека и экспериментальных жи |
||||||||
|
рогенное |
действие |
перекиси |
вотных |
обладают |
некоторые |
тяжелые |
|||
|
бензоила. |
|
|
|
|
металлы и их соединения: никель, |
||||
|
|
|
|
|
|
хром, |
бериллий, |
кадмий, |
кобальт, |
|
4.1.2.3.8. Прочие |
|
|
|
мышьяк, свинец, |
ртуть. Степень их |
|||||
|
|
|
канцерогенной активности и органы- |
|||||||
негенотоксичные соединения |
||||||||||
мишени во многом определяются рас |
||||||||||
|
|
|
|
|
|
|||||
Сахарин и лимонен — канцероге |
творимостью в тканевых жидкостях и |
|||||||||
ны, |
рассматриваемые |
вместе, |
по |
путях выведения из организма. В ча |
||||||
скольку действию их подвергаются ог |
стности, соединения никеля выводят |
|||||||||
ромные контингенты людей. Они тем |
ся с мочой и индуцируют у крыс опу |
|||||||||
не менее не представляют канцеро |
холи почек. Соединения хрома эли |
|||||||||
генной опасности для человека: саха |
минируются через желудочно-кишеч |
|||||||||
рин — вследствие ничтожно малой |
ный тракт и опухолей почек не вызы |
|||||||||
дозы, |
получаемой человеком, |
лимо |
вают. |
Большинство канцерогенных |
||||||
нен — вследствие видовой и половой |
производных этих металлов — полные |
|||||||||
специфичности |
его канцерогенного |
канцерогены, но некоторые являются |
||||||||
действия (только крысы-самцы). |
промоторами. Например, ацетат свин |
|||||||||
Сахарин вызывает |
рак |
мочевого |
ца и арсенит натрия — промоторы |
|||||||
пузыря у крыс и мышей при введении |
опухолей почек у крыс при инициа |
|||||||||
в течение всей их жизни в дозах 500 и |
ции нитрозосоединениями. В экспе |
|||||||||
1500 мг/кг в сутки. По формальным |
рименте показано, что основным ор |
|||||||||
признакам (индукция опухолей у двух |
ганом-мишенью для этой группы кан- |
|||||||||
218 |
|
|
|
|
|
|
|
|
|
|
>
церогенных веществ являются легкие, однако они могут вызывать опухоли и других органов: так, никель индуциру ет опухоли почек и саркомы на месте введения, бериллий — остеосаркомы, кадмий — лейкемии и опухоли яичка.
Ионы металлов электрофильны и способны взаимодействовать с нуклеофильными центрами макромоле кул. Металлы, попадающие в орга низм в виде порошка, подвергаются окислительно-восстановительным воздействиям, и их канцерогенность, видимо, связана также с образованием ионов.
В клетке катионы металлов спо собны образовывать активные формы кислорода, которые также могут уча ствовать в канцерогенном процессе. Для человека, как и в эксперименте, основным органом-мишенью для кан церогенных металлов и их соединений являются легкие. Помимо этой основ ной локализации, соединения никеля увеличивают риск заболеваемости ра ком полости носа и гортани, соедине ния кадмия — предстательной желе зы, шестивалентного хрома — полости носа, мышьяка — кожи и кроветвор ной ткани, свинца — желудка, почек и мочевого пузыря, а соединения рту ти — риск рака предстательной желе зы и почек.
4.1.3.2. Природные волокнистые и неволокнистые силикаты
Другой тип канцерогенеза связан с воздействием на организм природных и синтетических силикатов. Они раз личаются по структуре кристалличе ской решетки, содержанию ионов ме таллов, но общим является наличие окислов кремния. Канцерогенными свойствами обладают вещества, имею щие волокнистую структуру.
К волокнистым канцерогенным силикатам относятся различные виды асбеста, волокнистые цеолиты и син тетические волокна, полученные на основе кремнезема. Наиболее важным промышленным продуктом является
асбест. Предлагаемые в качестве его заменителей синтетические волокни стые силикаты также обладают канцерогенностью, хотя и меньшей, чем ас бест.
Среди природных асбестов наибо лее широко используется хризотил. Другие асбесты (амфиболы) в связи с сильной канцерогенностью запреще ны к применению или используются в очень небольших количествах. Глав ным потребителем хризотила является асбестоцементная промышленность.
Особенностью канцерогенных во локнистых силикатов является индук ция у людей и экспериментальных животных опухолей легких и мезотелиом плевры и брюшины. Канцеро генная опасность асбеста для человека несомненна, и МАИΡ включил его в группу веществ с доказанной канце рогенностью для людей (группа I). У рабочих, занятых на производствах по добыче, переработке и применению асбеста, риск заболевания раком лег ких и мезотелиомой плевры в не сколько раз выше, чем у лиц, не имеющих контакта с асбестом.
Среди волокнистых силикатов сле дует также упомянуть цеолит — эрионит, канцерогенность которого суще ственно выше, чем у асбеста.
Асбест обладает как инициирую щей, так и промотирующей активно стью. Являясь канцерогенным сам по себе, он усиливает канцерогенез в лег ких, индуцированный бенз(а)пиреном. Курящие рабочие асбестовой промышленности в 8 раз чаще заболе вают раком легких, чем некурящие, однако этого не отмечено для мезотелиом плевры.
Механизм канцерогенного дейст вия волокнистых силикатов ассоции руется с образованием активных форм кислорода. Предполагается две воз можности их образования:
• под влиянием ионов двухвалент ного железа, присутствующего в волокнах, кислород восстанав ливается до супероксида с по следующей дисмутацией до пе-
219
|
рекиси водорода и образованием |
го действия канцерогенов: при введе |
|||||||||||||||||||
|
гидроксилрадикала; |
|
|
|
|
нии канцерогена в первую неделю бе |
|||||||||||||||
• |
фагоциты |
активируются волок |
ременности преобладает |
эмбриоток- |
|||||||||||||||||
|
нами |
и |
производят |
|
активные |
сический эффект, во вторую — тера |
|||||||||||||||
|
формы |
кислорода, |
попадающие |
тогенный и в третью неделю — канце |
|||||||||||||||||
|
в клетки-мишени. |
|
|
|
|
рогенный |
эффект. |
В |
определенные |
||||||||||||
Силикатные |
волокна, |
однако, |
об |
дни |
беременности |
химическое соеди |
|||||||||||||||
нение может |
оказывать |
и |
тератоген |
||||||||||||||||||
ладают |
и теми |
свойствами, |
которые |
||||||||||||||||||
ное, и канцерогенное действие. |
|||||||||||||||||||||
отсутствуют |
у |
химических |
канцероге |
||||||||||||||||||
Некоторые |
ткани |
плода |
обладают |
||||||||||||||||||
нов: |
проникая |
в |
клетку |
в |
|
результате |
|||||||||||||||
|
высокой |
чувствительностью |
к канце |
||||||||||||||||||
фагоцитоза, |
они |
вызывают |
поврежде |
||||||||||||||||||
рогенам прямого действия. Например, |
|||||||||||||||||||||
ние веретена |
при |
митозе, фрагмента |
|||||||||||||||||||
ткани нервной системы плода в десят |
|||||||||||||||||||||
цию хромосом с образованием микро |
|||||||||||||||||||||
ки |
раз чувствительнее |
к |
канцероген |
||||||||||||||||||
ядер, |
а также |
непосредственное |
при |
||||||||||||||||||
ному действию нитрозометил- и нит- |
|||||||||||||||||||||
липание волокон к хромосомам. Воз |
|||||||||||||||||||||
розоэтилмочевины, чем нервная ткань |
|||||||||||||||||||||
можно, |
благодаря |
перечисленным |
|||||||||||||||||||
взрослого |
организма. Поскольку тка |
||||||||||||||||||||
свойствам |
канцерогенные |
минераль |
|||||||||||||||||||
ни |
эмбриона |
не |
обладают |
развитой |
|||||||||||||||||
ные волокнистые силикаты отличают |
|||||||||||||||||||||
системой |
монооксигеназ |
печени, ор |
|||||||||||||||||||
ся по |
своему |
действию |
от |
|
классиче |
||||||||||||||||
|
ганы эмбриона могут оказаться менее |
||||||||||||||||||||
ских |
химических негенотоксических |
||||||||||||||||||||
чувствительными |
к |
действию канце |
|||||||||||||||||||
соединений. Так, |
в некоторых тестах |
||||||||||||||||||||
рогенов, |
нуждающихся |
в |
метаболиче |
||||||||||||||||||
на мутагенность |
они |
дают |
положи |
||||||||||||||||||
ской активации. Выше |
говорилось о |
||||||||||||||||||||
тельный результат. Для индукции опу |
|||||||||||||||||||||
трансплацентарном действии на чело |
|||||||||||||||||||||
холи они не требуют введения вещест |
|||||||||||||||||||||
века диэтилстильбэстрола. |
|
||||||||||||||||||||
ва в течение всей жизни животных в |
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
больших дозах. Для них показана за |
4.1.4.2. |
Трансгенерационный |
|||||||||||||||||||
висимость доза — эффект. |
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
(мультигенерационный) |
|||||||||
4.1.4. |
|
|
Трансплацентарный |
канцерогенез |
|
|
|
|
|
||||||||||||
|
|
Это возникновение опухолей в ре |
|||||||||||||||||||
и |
|
трансгенерационный |
|
||||||||||||||||||
канцерогенез |
|
|
|
|
|
|
зультате |
канцерогенного |
воздействия |
||||||||||||
Эти два типа канцерогенеза рас |
на мужскую или женскую половую |
||||||||||||||||||||
клетку или обе половые клетки до их |
|||||||||||||||||||||
сматриваются |
|
здесь, |
поскольку |
они |
слияния, т. е. до образования зиготы. |
||||||||||||||||
иллюстрируют различия |
между канце |
|
Генотоксические |
канцерогены вы |
|||||||||||||||||
рогенами, нуждающимися и не нуж |
зывают и трансплацентарный, и муль |
||||||||||||||||||||
дающимися в метаболической актива |
тигенерационный канцерогенез. Неге |
||||||||||||||||||||
ции, а также между генотоксическими |
нотоксические |
|
(эпигенетические) |
||||||||||||||||||
инегенотоксическими канцероге канцерогены, по-видимому, не спо
нами. |
собны индуцировать ни ту, ни другую |
|
|
разновидности канцерогенеза. В этом |
|
4 . 1 . 4 . 1 . Трансплацентарный |
отношении показательны результаты |
|
опыта с ДДТ, который вводили мы |
||
канцерогенез |
||
шам в нескольких дозах на протяже |
||
|
||
Это обычный химический канце |
нии 6 поколений, т. е. 5 поколений |
|
рогенез, при котором имеет место |
мышей получали канцероген не толь |
|
прямое действие канцерогена на со |
ко на протяжении всей жизни, но и |
|
матическую клетку-мишень плода; |
трансплацентарно. Повышения часто |
|
достигнуть этой клетки-мишени кан |
ты опухолей печени от поколения к |
|
цероген может, лишь преодолев пла |
поколению, чего можно было бы ожи |
|
центарный барьер. Имеет место ста- |
дать при трансплацентарном и муль- |
|
диоспецифичность трансплацентарно |
тигенерационном канцерогенезе, от- |
220
мечено не было. ДДТ является типич ным слабогенотоксическим канцеро геном: в большинстве тестов на гено токсичность он давал отрицательные или противоречивые результаты. При чиной отсутствия трансплацентарного эффекта была, вероятнее всего, недос таточная доза канцерогена. Выше го ворилось, что негенотоксический кан цероген для индукции опухолей необ ходимо вводить в высоких дозах и длительно. Очевидно, что срок 1,5 нед беременности, в течение которых ДДТ мог проявить канцерогенные свойства при трансплацентарном введении, со вершенно недостаточен для индукции опухолей.
Известна и наследственная переда ча в эксперименте процесса инициа ции. На самца действуют генотоксическим канцерогеном (например, ДМБА), а на потомков, полученных от этого самца, наносят промотор ТФА, в результате чего возникают па пилломы в значительно большем ко личестве, чем при нанесении ТФА на кожу контрольных мышей, родители которых не подвергались действию канцерогена. Эта ситуация в какой-то мере напоминает представления Knudson (1975) о двухмутационном происхождении наследственной ретинобластомы: в половой клетке родите ля, а следовательно, и во всех клетках его детей, в том числе и в клетках сет чатой оболочки, имеется лишь один мутированный ген. Мутация второго гена, необходимая для развития ретинобластомы, происходит в клетке ре тины на протяжении последующей жизни.
4.1.5. Классификации химических канцерогенов по степени канцерогенной опасности для человека (так называемые гигиенические классификации)
Суммарная оценка канцерогенной опасности (реальной или потенциальной) химических соединений для че
ловека производится на основе ком плексного анализа эпидемиологиче ских данных, результатов эксперимен тальных испытаний на животных и результатов краткосрочных тестов.
Из гигиенических классификаций наибольшей известностью пользуется классификация Международного аген тства по изучению рака (МАИР), в которой предусмотрены 4 группы.
Группа 1. Агент (или смесь аген тов) канцерогенен для человека. Дос таточные доказательства канцероген ности для человека (установлена при чинная связь между воздействием агента и возникновением злокачест венных опухолей у человека).
Химические вещества и другие факторы, канцерогенность которых для человека доказана, — группа 1 по классификации МАИР (частично материал представлен в главе 1, табл. 1.4, 1.5)
Агенты и группы агентов
1.Афлатоксины, встречающиеся в природе.
2.4-Аминобифенил.
3.Мышьяк и соединения мышьяка.
4.Асбест.
5.Азатиоприн.
6.Бензол.
7.Бензидин.
8.Бериллий и соединения бериллия.
9.N,N-бис(2-хлорэтил)-2-нафтила- мин (хлорнафазин).
10.Бис(хлорметиловый)эфир и хлорме- тил-метиловый эфир.
11.1,4-Бутандиол диметансульфонат (бусульфан, милеран).
12.Кадмий и соединения кадмия.
13.Хлорамбуцил.
14.1-(2-хлорэтил)-3-(4-метилциклогек-
сил)-1-нитрозомочевина |
(сему- |
стин). |
|
15.Соединения хрома.
16.Циклоспорин.
17.Циклофосфамид.
18.Диэтилстильбэстрол.
19.Вирус Эпштейна—Барр.
20.Эрионит.
21.Этилена оксид.
22.Этопозид в сочетании с цисплатином и блеомицином.
23.Гамма-излучение.
24.Helicobacter pylory.
221
25.Вирус гепатита В.
26.Вирус гепатита С.
27.Вирус иммунодефицита человека, тип 1.
28.Вирус папилломы человека, тип 16.
29.Вирус папилломы человека, тип 18.
30.Т-клеточный лимфотропный вирус человека, тип 1.
31.Мелфалан.
32.8-Метоксипсорален (метоксален) плюс ультрафиолетовое излучение.
33.МОРР и другая комбинированная химиотерапия, включающая алкилирующие агенты.
34.Горчичный газ, иприт (горчичный сульфид).
35.2-Нафтиламин.
36.Нейтроны.
37.Соединения никеля.
38.Эстрогенотерапия постменопаузальная.
39.Эстрогены нестероидные.
40.Эстрогены стероидные.
41.Opistorchis viverrini.
42.Оральные контрацептивы комбини рованные.
43.Оральные контрацептивы некомби нированные.
44.Фосфор-32 в виде фосфата.
45.Плутоний-239 и продукты его рас пада (может содержать плутоний240 и другие изотопы) в виде аэро золей.
46.Радиоактивные соединения йода, короткоживущие изотопы, включая йод-131, как последствие аварий в атомных реакторах и при взрыве ядерной бомбы (экспозиция в дет стве).
47.Радионуклиды, излучающие α-час тицы, интеркорпоральная кумуля ция.
48.Радионуклиды, излучающие β-час- тицы, интеркорпоральная кумуля ция.
49.Радий-224 и продукты его распада.
50.Радий-226 и продукты его распада.
51.Радий-228 и продукты его распада.
52.Радон-222 и продукты его распада.
53.Schistosoma haematobium.
54.Двуокись кремния (кремнезем) кристаллическая (вдыхаемая в виде кварца или кристобалита, источник поступления в организм связан с профессией).
55.Солнечная радиация.
56.Тальк, содержащий асбестоподобные волокна.
57.Тамоксифен.
58.2,3,7,8-Тетрахлородибензо-пара-ди- оксин.
59.Тиотефа (тиофосфамид).
60.Торий-232 и продукты его распада, введенные внутривенно в виде кол лоидной взвеси диоксида тория232.
61.Треосульфан.
62.Хлористый винил.
63.Рентгеновское (X) и гамма(гамма)-излу- чение.
Смеси
1.Алкогольные напитки.
2.Обезболивающие смеси, содержа щие фенацетин.
3.Бетель с табаком.
4.Каменноугольные смолы.
5.Каменноугольные дегти.
6.Минеральные масла, необработан ные или умеренно обработанные.
7.Соленая рыба.
8.Сланцевые масла.
9.Сажи.
10.Продукты табака, бездымные.
11.Табачный дым.
12.Древесная пыль.
Экспозиция
1.Производство алюминия.
2.Аурамин, производство аурамина.
3.Производство и ремонт обуви.
4.Газификация каменного угля.
5.Производство кокса.
6.Мебельное производство.
7.Разработка гематита (подземная) с экспозицией к радону.
8.Литье железа и стали.
9.Производство изопропанола (силь нокислотный процесс).
10.Фуксин (красная анилиновая крас ка), производство фуксина.
11.Профессия маляра (профессиональ ная экспозиция).
12.Резиновая промышленность.
13.Сильнокислотные неорганические частицы, содержащие серную ки слоту (профессиональная экспози ция).
Группа 2. Степень доказательности канцерогенности для человека, с од ной стороны, является почти доста точной, а с другой — эпидемиологи ческие данные отсутствуют, но име ются доказательства канцерогенности для животных. Группа 2 разделена на две подгруппы — 2А (вероятный кан-
222
цероген человека) и 2В (возможный канцероген человека). Степень дока зательности канцерогенности для че ловека в подгруппе 2А выше, чем в подгруппе 2В.
Химические вещества и другие факторы, канцерогенность которых для человека является вероятной (группа 2А
по классификации МАИР)
Агенты и группы агентов
1.Акриламид.
2.Адриамицин.
3.Андрогенные (анаболические) сте роиды.
4.Азацитидин.
5.Бенз(а)антрацен.
6.Бензидинсодержащие красители.
7.Бенз(а)пирен.
8.Бис-хлорэтил нитрозомочевина.
9.1,3-Бутадиен.
10.Каптафол.
11.Хлорамфеникол.
12.α-Хлорированные толуолы (бензальхлорид, бензотрихлорид, бензилхлорид и бензоилхлорид (комби нированная экспозиция).
13.1-(2-хлорэтил)-3-циклогексил-1- нитрозомочевина.
14.Пара-хлоро-орто-толуидин и его сильнокислотные соли.
15.Хлорзотоцин.
16.Цисплатин.
17.Clonorchis sinensis.
18.Дибенз(а,h)антрацен.
19.Диэтилсульфат.
20.Диметилкарбамоила хлорид.
21.1,2-Диметилгидразин.
22.Диметила сульфат.
23.Эпихлоргидрин.
24.Этилена дибромид.
25.Ν-этил-N-нитрозомочевина.
26.Этопозид.
27.Формальдегид.
28.Глицидол.
29.Вирус папилломы человека, тип 31.
30.Вирус папилломы человека, тип 33.
31.2-Амино-3-метилимидазо(4,5-f)хи- нолин.
32.Герпес-вирус саркомы Капоши / герпес-вирус 8 человека.
33.5-Метоксипсорален.
34.4,4'-Метилен бис(2-хлоранилин).
35.Метил меиансульфонат.
36.N-метил-N'-нитро-N-нитрозогуа- нидин.
37.Ν-метил-М-нитрозомочевина.
38.Азотистый иприт.
39.Ν-нитрозодиэтиламин.
40.Ν-нитрозодиметиламин.
41.Фенацетин.
42.Прокарбазина гидрохлорид.
43.Стирен-7,8-оксид.
44.Тенипозид.
45.Тетрахлорэтилен.
46.Ортотолуидин.
47.Трихлорэтилен.
48.1,2,3-Трихлорпропан.
49.Трис(2,3-дибромпропил) фосфат.
50.Ультрафиолетовая радиация А.
51.Ультрафиолетовая радиация В.
52.Ультрафиолетовая радиация С.
53.Винила бромид.
54.Винила фторид.
Смеси
1.Креозоты.
2.Выхлоп дизельного двигателя.
3.Горячий напиток из листьев дерева
Ilex paraguariensis (родина — Пара гвай и Аргентина).
4.Инсектициды, не содержащие мышьяк (профессиональная экспо зиция в распыленном виде и путем аппликации).
5.Полихлорированные бифенилы.
Экспозиция
1.Производство художественного стекла, стеклянных контейнеров и изделий из прессованного стекла.
2.Профессия парикмахера (профес сиональная экспозиция).
3.Очистка нефти (профессиональная экспозиция).
4.Солнечные лампы и солнечные кровати, их использование.
Группа 3. Агент не может быть классифицирован с точки зрения его канцерогенности для человека: неаде кватные доказательства канцероген ности для человека и неадекватные или ограниченные доказательства канцерогенности для животных.
Группа 4. Агент, вероятно, не кан церогенен для человека: доказательст ва отсутствия канцерогенности для человека и животных или неадекват ные доказательства канцерогенности для человека с отсутствием канцеро генности у животных и отрицательны ми поддерживающими данными (от-
223
сутствие |
генотоксичности, |
|
тератоген- |
человека. В зависимости от принад |
||||||||||||||||||
ности и др.). |
|
|
|
|
|
|
лежности агентов к той или другой |
|||||||||||||||
В |
отечественной |
классификации |
категории |
предусматриваются условия |
||||||||||||||||||
пестицидов, весьма сходной с класси |
их практического применения. |
|
||||||||||||||||||||
фикацией МАИР, |
использована тер |
Три |
категории |
предусматриваются |
||||||||||||||||||
минология, |
применяемая |
в токсико |
классификацией |
Европейского сооб |
||||||||||||||||||
логии: "классы" вместо "групп"; введен |
щества: 1 — канцероген человека; 2 — |
|||||||||||||||||||||
дополнительный подкласс |
|
2С; указа |
весьма вероятный канцероген челове |
|||||||||||||||||||
ны ограничения в применении хими |
ка; 3 — возможный канцероген чело |
|||||||||||||||||||||
ческих агентов в зависимости от их |
века. На этикетках первых двух кате |
|||||||||||||||||||||
принадлежности к тому или другому |
горий |
указывается: |
"Может |
вызвать |
||||||||||||||||||
классу. Так, химические соединения I |
рак", на этикетках 3-й категории: |
|||||||||||||||||||||
класса опасности, как правило, не ре |
"Возможен |
риск |
необратимого |
эф |
||||||||||||||||||
комендуются для использования в на |
фекта". |
|
|
|
|
|
|
|
|
|
|
|||||||||||
родном хозяйстве, и их ограниченное |
|
|
|
|
|
* * * |
|
|
|
|
|
|||||||||||
применение возможно только специа |
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
листами и лишь в случаях крайней не |
Основные различия между гено- |
|||||||||||||||||||||
обходимости при условии сведения к |
||||||||||||||||||||||
минимуму |
опасности |
для |
работаю |
токсическими |
|
и |
эпигенетическими |
|||||||||||||||
щих, населения и окружающей среды; |
(негенотоксическими) |
канцерогенами |
||||||||||||||||||||
розничная продажа запрещается. Пре |
заключаются в механизмах их канце |
|||||||||||||||||||||
параты |
II |
класса |
опасности |
могут |
рогенного |
действия. |
|
Если |
геноток |
|||||||||||||
быть |
использованы только специали |
сический механизм канцерогенеза яв |
||||||||||||||||||||
стами при условии строгой регламен |
ляется |
практически |
|
универсальным |
||||||||||||||||||
тации и ограничения их применения |
для всех видов, включая человека, то |
|||||||||||||||||||||
от умеренного в подклассе 2С вплоть |
важной |
особенностью |
эпигенетиче |
|||||||||||||||||||
до запрещения в подклассе 2А. Их |
ских |
канцерогенов |
является |
видовая, |
||||||||||||||||||
розничная продажа разрешается толь |
линейная, |
половая |
|
специфичность |
||||||||||||||||||
ко лицам, |
прошедшим |
подготовку. |
механизмов их канцерогенного дейст |
|||||||||||||||||||
Препараты III и IV классов опасности |
вия. Принципиальным различием ме |
|||||||||||||||||||||
могут применяться без ограничений (с |
жду |
генотоксическими |
и |
негеноток |
||||||||||||||||||
точки зрения их канцерогенной опас |
сическими |
канцерогенами |
является |
|||||||||||||||||||
ности) в соответствии с действующи |
то, что первые оставляют след в гено |
|||||||||||||||||||||
ми санитарными нормами и инструк |
ме клетки в виде активации того или |
|||||||||||||||||||||
циями. |
|
|
|
|
|
|
|
|
иного специфического для этого кан |
|||||||||||||
"Перечень |
веществ, |
|
продуктов, |
церогена онкогена |
или инактивации |
|||||||||||||||||
производственных |
процессов, |
быто |
антионкогена. Если в системе ини |
|||||||||||||||||||
вых и природных факторов, лекарст |
циация |
— |
промоция |
действовали |
||||||||||||||||||
венных препаратов" (М., 1999), яв |
один инициатор и ряд промоторов, то |
|||||||||||||||||||||
ляющийся |
гигиеническим |
нормати |
во всех случаях независимо от харак |
|||||||||||||||||||
вом для РФ, делит их на группы: 1) с |
тера промотора будет иметь место |
|||||||||||||||||||||
доказанной канцерогенностью для че |
один и тот же след в геноме — актива |
|||||||||||||||||||||
ловека и 2) вероятно канцерогенные |
ция |
онкогена, |
специфического |
для |
||||||||||||||||||
для человека. |
|
|
|
|
|
|
данного генотоксического |
канцероге |
||||||||||||||
В классификации Агентства по ох |
на. При обратной ситуации — различ |
|||||||||||||||||||||
ные инициаторы при одном и том же |
||||||||||||||||||||||
ране |
окружающей |
среды |
США (USA |
|||||||||||||||||||
промоторе |
— |
в |
геноме будут обнару |
|||||||||||||||||||
Environmental |
Protection Agency) |
име |
||||||||||||||||||||
жены различные метки, соответствую |
||||||||||||||||||||||
ется 6 категорий: А — канцероген че |
||||||||||||||||||||||
щие примененным генотоксикантам. |
||||||||||||||||||||||
ловека, В1 и В2 — вероятные канце |
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
рогены человека, С — возможный |
Принято |
считать, |
что |
механизм |
||||||||||||||||||
канцероген человека, D — некласси |
канцерогенного |
действия |
генотокси |
|||||||||||||||||||
фицированный агент, Ε — отсутствие |
ческих |
канцерогенов |
является беспо |
|||||||||||||||||||
доказательств |
канцерогенности |
для |
роговым, т. е. теоретически достаточ- |
|||||||||||||||||||
224