

Мальабсорбція при схо вуглеводів (Хоффманн г., Гречаніна о.Я., 2005)
Порушення |
Індекс Мак-Кьюсіка |
Фермент |
Основний субстрат |
Непереносимість сахаридів І |
222 900 |
β- фруктофуранозидаза / ізомальтаза |
сахароза |
Непереносимість сахаридів ІІ- природжена алактазія |
223 000 |
комплекс β-глюкозидази- лактази, глікозілцерамідази |
лактоза |
Непереносимість сахаридів ІІІ - недостатність лактази у дорослих |
223 100 |
комплекс β-глюкозидази - лактази, глікозілцерамідази |
лактоза |
Недостатність трегалази |
275 360 |
трегалаза |
трегалоза (гриби) |
Не виявлено |
154 360 |
глюкоамілаза (мальтаза) I и II |
|
Непереносимість сахаридів І. Недостатність β-фруктофуранозидази/ ізомальтази є нечастою причиною інфантильної діареї, що стає очевидною при включенні сахарози в дієту. Існує кілька різних механізмів, що впливають на виникнення ферментативної недостатності, серед яких порушена секреція, аномальне утворення складок, недостатня каталітична активність і підвищена деструкція.
У клінічному відношенні глюкозо-галактозна мальабсорбція подібна до недостатністі β-фруктофуранозидази/ізомальтази і з раннього дитячого віку характеризується важкою формою водянистих (осмотичних) випорожнень. Своєчасна елімінація з дієти глюкози й галактози та дієтотерапія на основі фруктози дає високий лікувальний ефект. Встановлено, що цей вид мальабсорбції пов'язаний з дефектом у натрієво-глюкозному котранспортері SGLT1, що є результатом мутацій у гені SLC5A1 розчиненого транспортеру.
Непереносимість сахаридів ІІ. Природжена недостатність лактази зустрічається надзвичайно рідко. Майже у всіх випадках недостатність лактази у дітей раннього дитячого віку й у дітей молодшого дитячого віку є результатом інфекції та втрати дозрілої щіткової облямівки. Природжена непереносимість лактози, очевидно, відрізняється від природженої недостатності лактази. При природженій нестерпності лактози спостерігаються надмірне поглинання лактози шлунком (що приводить до лактоземії й лактозурії), блювота, затримка розвитку, порушення функції печінки й нирковий синдром Фанконі. Природжена недостатність лактази може мати летальний результат у випадку її невиявлення та у випадку її нелікування шляхом елімінації лактози з дієти. Цікаво, що лактоза добре переноситься по досягненні віку 6 місяців. Що лежить в основі цього захворювання, залишається нез'ясованим.
Непереносимість сахаридів ІІІ. Недостатність лактази у дорослих являє собою поліморфізм, що часто зустрічається. По суті експресія лактази в кишечнику, що знижується в дитячому віці, є звичайним станом, що відбиває роль припинення годування грудним молоком у дітей, що перебороли грудний вік. У деяких регіонах Північної Європи у дітей, що перебороли грудний вік, непоквашене молоко залишається важливим дієтичним компонентом. Здатність засвоювати молоко має безсумнівну користь, тому серед індивідів, що проживають у цих регіонах, непереносимість лактози спостерігається лише в меншостей. Можливо, традиція пити непоквашене молоко виникла саме через те, що з віком випадкова мутація перешкоджає звичайному зниженню активності лактази. Активність лактази в кишечнику залежить від сумарної експресії обох алелей. Недавні дослідження показують, що розходження в експресії лактази перебувають не в самій послідовності, що кодує ген, а, імовірно, у діючому в цис-положенні регуляторному елементі.
Мальабсорбція глюкози-галактози. Непереносимість моносахаридів є результатом порушення активного транспорту глюкози й галактози в слизовій оболонці тонкої кишки, що обумовленого мутаціями (більше 30) гену SCLT. Захворювання успадковується аутосомно-рецесивно. У результаті виникає дефект специфічного білка-переносника, що органічно пов'язаний з мембранними структурами ентероциту. Аналогічний дефект виявляють і в ниркових канальцях, що підтверджує єдність функціональних транспортних блоків слизової оболонки тонкої кишки та канальців нирок. Можливі варіанти захворювання з переважним ураженням кишечника або нирок.
Глюкозо-галактозна мальабсорбція проявляється проносами вже в перші 2-4 дня життя. Дитина близько 10-20 разів у добу виділяє водянисті, що нагадують сечу, випорожнення з кислим запахом, що містять велику кількість глюкози й галактози, іноді зі слизом. Швидко розвивається дегідратація, спостерігається сильна спрага, може бути підвищення температури тіла як прояв вододефіцитної дегідратації. Живіт роздутий, може бути блювота. Незважаючи на дегідратацію, зберігається поліурія. При припиненні молочного харчування, після парентеральної регідратації симптоми проходять.
З віком ступінь клінічних проявів звичайно пом'якшується, важлива роль в адаптації до глюкози, що не всмокталася, належить кишковій мікрофлорі, яка метаболізує моносахариди. При цьому призначення антибіотиків, що пригнічують нормальну кишкову мікрофлору, приводить до погіршення клінічної симптоматики захворювання. На все життя зберігається погана переносимость молока й солодощів.
У сечі та калі таких хворих виявляють глюкозу й галактозу. Глюкозурія визначає необхідність виключення цукрового діабету, але, на відміну від останнього, рівень глюкози в крові знижений. Цукрова крива з навантаженням глюкозою має плоский вигляд.
Захворювання варто диференціювати з галактоземією, що обумовлена порушенням метаболізму галактози в печінці, і з природженою формою лактазної недостатності.
Мальабсорбція фруктози. Нечаста генетично детермінована форма порушеного усмоктування, пов'язана з порушенням п'ятого шляху транспорту глюкози (синтезу глюкози із фруктози в стінці кишечника з наступним транспортом). При цьому інші шляхи транспорту глюкози через глюкозо- галактозний або глюкозо-натрієвий канали не порушені.
Клінічні симптоми з'являються після вживання соків і фруктів, що містять велику кількість фруктози: апельсини, яблука, вишні, черешні. Симптоматика залежить від кількості вжитої фруктози. Фруктоза, що не всмокталася, маючи осмотичну дію, викликає водянисту діарею, метеоризм, іноді блювоту та коликоподібний біль у животі.
Вживання фруктів або соків, що містять суміш глюкози й фруктози (банани), не викликає значимого градієнта концентрації між порожниною кишки й внутрішнім середовищем. Тому при прийманні в їжу таких фруктів осмотичної діареї не виникає. Важливе діагностичне значення має вказівка на погану переносимість фруктів і соків. Якщо прямих вказівок на це не має, корисну інформацію може дати ведення харчового щоденника з позначкою про характер і час виникнення патологічних симптомів. При підозрі на мальабсорбцію фруктози проводять цукрову криву або водневий тест із фруктозою, плоский вид кривої та наростання концентрації водню після прийому фруктози підтверджують діагноз.
Диференціальний діагноз проводять із фруктоземією, для якої характерне збільшення печінки, виявлення фруктози в сечі та зниження активності печіночного ферменту фруктозо-1-фосфатальдолази.
Первинна мальабсорбція солей жовчних кислот – аутосомно-рецисивне захворювання, пов'язане з порушенням синтезу білка-транспортера солей жовчних кислот у клубовій кишці. Генетичні мутації виявляють при цьому в локусі SLC10А2 на хромосомі 13q33. Жовчні кислоти, що не всмокталися, стимулюють кишкову секрецію і моторику, розвивається діарея секреторного типу, що має дуже стійкий характер, не купується при голодуванні, і переведенні дитини на парентеральне харчування. Захворювання проявляється з перших місяців життя, симптоматика підсилюється при переведенні на штучне вигодовування. У добу дитина може втрачати з випорожненнями більше 900 мг/ м2, швидко прогресує гіпотрофія, відставання у розвитку. Порушення всмоктування жиророзчинних вітамінів (зокрема, вітаміну К) може проявлятися геморагічним висипом на шкірі, іноді спостерігають набряки аж до анасарки. З часом розвивається вторинний остеопороз. Характерне також збільшення печінки. У копрограмі виявляють стеаторею за рахунок жирних кислот. У крові знижений рівень холестерину, може бути гіпопротеїнемія. Характерне підвищення циркулюючих імунних комплексів і зниження комплементу.
Мальабсорбція триптофану (хвороба Хартнупа) являє собою аутосомно- рецесивне порушення, що може бути клінічно безсимптомним. У патологічний процес залучений транспорт декількох нейтральних амінокислот у кишечнику та у нирках, однак триптофан, найімовірніше, є однією з найбільш потребуючих обмеження прийому. У тому випадку, якщо застосування ніацину в дієті також не відповідає вимогам, недостатність нікотинової кислоти й нікотинаміду викликає появу висипу (пелагри), світлочутливості, емоційної неврівноваженості й атаксії. При важких випадках захворювання ця недостатність переростає в енцефалопатію із властивими їй делірієм і приступами. При цьому може спостерігатися підвищення рівня індолів у випорожненнях та індикану в сечі, що відображає вплив кишкових бактерій на неабсорбований триптофан. Постановка діагнозу передбачається шляхом виявлення високих рівнів нейтральних амінокислот у сечі: аланіну, серину, треоніну, аспарагіну, глутаміну, валіну, лейцину, ізолейцину, фенілаланіну, тирозину, триптофану, гістидину й цитруліну. Рівні цих амінокислот у плазмі крові виявляються нормальними або злегка зниженими.
Як уже було зазначено, основним місцем всмоктування Са в організмі є кишечник. І хоча депо Са – кісткова тканина, його вміст у м'яких тканинах є обов'язковим, що обумовлено функцією даного елементу в організмі. У різних відділах кишечника мінеральний склад кишкової стінки має достовірну різницю. Так, у верхніх відділах (дванадцятипала і початковий відділ тонкої кишки) вміст Са не перевищує 0,82-0,85 %, тоді як у прямій кишці він досягає 1,7 %, а в сліпій – 2,4 %. Слід підкреслити, що вміст Са в тканинах людини істотно змінюється протягом життя: цей показник значно вищий у літньому віці.
Механізми, які беруть участь у підтримці нормальної концентрації іонізованого позаклітинного Са, регулюють його абсорбцію у ШКТ, екскрецію нирками і процеси обміну в кістках. Одна з основних функцій Са в організмі – забезпечення скорочення м'язів, у тому числі гладкої мускулатури травного тракту. При цьому моторна функція кишечника регулюється двома шляхами – нервовим та гуморальним.
Одним із основних патологічних механізмів розвитку функціональних і органічних захворювань кишечника є надмірне скорочення гладеньких м'язів кишкової стінки, що викликає абдомінальний біль, а у деяких випадках і запори (внаслідок порушення моторики кишечника – посилення непропульсивних і сегментарних рухів та зниження пропульсивної активності з підвищенням тонусу сфінктерів). Мабуть, у цьому випадку має місце локальне порушення кальцієвого гомеостазу в міоцитах кишкового тракту – підвищена концентрація Са ²+ у клітині і порушений вихід його з міоцитів.
Відомо, що Са сироватки крові знаходиться у динамічній рівновазі з солями цього іона, що міститься в кістках. Таким чином, порушення моторики кишечника, які супроводжуються надлишковим споживанням позаклітинного Са, вимагають поповнення Са з кісткового депо за рахунок процесів ремоделювання з поступовим збіднінням кальцієвих запасів при тривалому перебігу патології кишечника. І, навпаки, однією з причин гіпокальціємії, поряд зі зниженням абсорбції Са, є порушення моторики та тонусу кишечника. Таким чином, хронічна гіпокальціємія і гіповітаміноз D, а також порушення всмоктування Са в кишечнику можуть бути причиною розвитку остеомаляції та остеопорозу.
Синдром де Тоні-Дебре-Фанконі (проксимальна тубулопатія) характеризується розвитком проксимальної тубулопатії, вітамін D- резистентного рахіту, гіпотрофії вже на 1- му році життя хворої дитини. Стійка анорексія та, як наслідок її, псевдонепрохідність кишечника, що проявляється запорами, змінюються діареєю. Іноді маніфестація захворювання відбувається дуже рано – на 1-му місяці життя, супроводжуючись приступами дегідратації, поліурії, метаболічного ацидозу, і призводить до летальних наслідків на висоті кетоацидотичної коми. Пізніше може приєднатися мультиорганна патологія у вигляді печінкової недостатності, цукрового діабету, міоклоній, зовнішньої офтальмоплегії, глухоти й сліпоти. Хронічна гіпокальціємія і гіповітаміноз D викликають розвиток остеомаляції та остеопорозу.
Відповідно до гіпотези де Тоні, в основі патогенезу захворювання лежить ферментативний дефект у циклі Кребса й, отже, енергетична недостатність канальців епітелію. Останні дослідження показали, що розвиток синдрому пов'язаний із мутацією мітохондріального геному, що супроводжується ізольованою або поєднаною недостатністю комплексів дихального ланцюга ІІІ, ІV, ІІІ+ІV, причому переважна мутація мітохондріальної ДНК (мтДНК) – у клітинах ниркових канальців. Біохімічно визначається генералізована нирково-тубулярна дисфункція з масивним виведенням амінокислот, глюкози, фосфатів, уратів і бікарбонатів.
Хвороба Гіршпрунга (природжений кишковий агангліоз, агангліонічний мегаколон, анальний агангліоз) характеризується відсутністю інтрамуральних парасимпатичних гангліїв на відрізку товстої кишки різної довжини, проксимально від області ануса. Захворювання зустрічається в одного з 8000 живонарождених, причому в хлопчиків частіше.
У більшості випадків хвороба Гіршпрунга зустрічається спорадично. Тільки в 4 % випадків вона буває сімейним захворюванням з багатофакторним, модифікованим за статтю типом успадкування з низьким порогом для осіб чоловічої статі. Якщо в родині виявлений хворий із коротким сегментом кишечника (ректосигмоїдний відділ або тільки пряма кишка), то має місце особливо високий ризик захворювання для осіб чоловічої статі, тоді як у родинах, члени яких страждають агангліозом на довгому відрізку кишки, це розходження зникає. Ризик повторюваності захворювання для близьких родичів пробанда становить 10 % при варіанті довгого сегменту. У деяких випадках можливий розвиток аутосомно-домінантного типу з варіабельною пенетрантністю.
Клінічна картина характеризується стійкими закрепами (100 % випадків) і динамічною кишковою непрохідністю. В 45 % випадків відзначається блювота, у 85 % випадків – здуття живота. Відділ кишечника вище агангліозної ділянки розширюється, стінки гіпертрофуються, розвивається мегаколон, що може ускладнится ентероколітом і перфорацією кишки. Калова інтоксикація може викликати жирову дистрофію печінки.
До патогномонічних ознак захворювання відноситься відсутність гангліонарних клітин у міжм’язовому сплетенні кишечника. Зона ураження звичайно обмежується областю сигмовидної або прямої кишки. При залученні в патологічний процес сліпої кишки використовується термін "варіант ураження довгого сегменту кишечника"; цей стан зустрічається рідко (усього 6-21 % всіх випадків). Ще більш рідко реєструється множинна сегментарне ураження всієї товстої та частково прямої кишки (так званий стрибкоподібний агангліоз). Термін "тотальний агангліоз товстої кишки" використовується при залученні в патологічний процес всього товстого кишечника та різних відділів тонкої кишки. Цей стан зустрічається в 8 % хворих.
Труднощі пренатальної діагностики за допомогою ферментного аналізу можуть бути пов'язані з недостатньою виразністю захворювання в ІІ триместрі вагітності. В 70-80 % випадків хвороба діагностується тільки в неонатальному періоді. Внутрішньоутробна ідентифікація кишкової непрохідності, пов'язаної з хворобою Гіршпрунга, повинна виключати розвиток ускладнень внаслідок пізнього встановлення діагнозу. До таких ускладнень відносяться ентероколіт, перфорація сліпої кишки, обструктивна уропатія, дефіцит живильних речовин в організмі й заворот кишок.
У випадку двох останніх спадкових захворюваннях (синдром де Тоні-Дебре-Фанконі та хвороба Гіршпрунга) основними патогенетичними чинниками розвитку вторинного остеопорозу є хронічна гіпокальціємія і гіповітаминоз D, а також порушення всмоктування Са унаслідок відсутності нормальної моторики кишечника.
Однак, має значення також інший механізм формування кальцієвого дисбалансу, наприклад – при хронічному коліті або синдромі роздратованого кишечника.
Са повинен постійно надходити в організм для поповнення фізіологічних потреб. Добове надходження Са з їжею становить 0,6-1,5 г, абсорбується його близько 50-75 %. При цьому встановлено, що всмоктування Са найбільш інтенсивно відбувається в початкових відділах тонкого кишечника. Суттєво впливає на всмоктування Са в кишечнику присутність речовин, які зв'язують Са і зменшують його абсорбцію.
Інтенсивність всмоктування Са залежить від багатьох факторів, до яких відносяться: функціональний стан органів травного тракту, характер зв'язку Са в харчових продуктах і їх кількість, забезпеченість організму вітаміном D, наявність жовчі в дванадцатипалій кишці, тип з'єднання Са (важкорозчинні або легкорозчинні солі), водний розчин або харчові продукти (швидше Са всмоктується з розчинів), концентрація і доза Са, наявність інших сполук (білки, глюкоза, галактоза, фруктоза і сахароза підвищують засвоєння Са); переважання в харчовому раціоні лужних елементів уповільнює, а переважання кислої їжі підсилює резорбцію Са.
Виявлена закономірність вказує на те, що інтенсивність всмоктування Са в кишечнику значно підвищується при надходженні вітаміну D і жовчі. У той же час, жовчні кислоти відіграють важливу роль у метаболізмі самого вітаміну D. У зв'язку з цим кишечник можна розглядати як депо мінеральних солей, поповнення котрого здійснюється за рахунок екзогенного і ендогенного Са, що виділяється залозами травного каналу (за добу утворюється більше 8 літрів сольового розчину).
Більша частина ендогенного Са знову резорбується в кишечнику і тільки частина його виводиться з організму. При цьому Са виводиться з калом (56-97 % всього Са); на екскрецію нирками припадає лише 10-30 %. У той же час було встановлено, що виведення ендогенного Са з організму підтримується на досить високому рівні в умовах навіть його негативного харчового балансу. Так у людини, яка споживає лише 100 мг Са, середня добова його втрата з калом може протягом декількох днів залишатися рівною 300 мг. Ось чому кишечник не можна розглядати тільки як порожнину, з якої організм постійно поповнює свої запаси Са і забезпечує виведення з калом солей, які не всмоктатися. Залозистий апарат кишечника є важливою ланкою в механізмі регуляції кальцієвого обміну.
Наведені дані стали підставою для проведення дослідження, метою котрого було вивчення стану кальцієвого обміну та кісткового метаболізму у хворих із функціональним (синдром роздратованого кишечника – СРК) і органічним (хронічний коліт) захворюваннями кишечника. Так, у дослідженнях А.Б. Андруша (2006 р.) було показано, що у хворих із СРК і хронічним колітом має місце негативна динаміка кальцієвого гомеостазу. На показники кальцієвого обміну впливали: вік хворих, тривалість анамнезу, нозологічна форма (хронічний коліт чи СРК). Крім того було встановлено, що зміни в показниках кальцієвого обміну визначали вираженість больового синдрому і тип дискінезії товстої кишки.
Отже, кальцієвий дисбаланс при хронічній патології кишечника може бути не тільки наслідком ураження кишечника, але і в результаті змін у гомеостазі Са формується клініка хронічного коліту та СРК.
Проведеною роботою було показано, що зміни до кальцієвого гомеостазу призводять до порушення кісткового ремоделювання навіть у хворих із функціональними захворюваннями кишечника, а саме СРК. Дані результати були отримані при проведенні ультразвукового денситометричного дослідження, а також при вивченні вмісту маркерів кісткової резорбції. І хоча хворих з остеопенічного станами при наявності СРК було менше (близько 12 %), ніж у групі пацієнтів із хронічним колітом (у 19 % діагностувалася остеопенія, а в 27 % – остеопороз різного ступеня виразності), це підтверджує їхню роль у формуванні даного ускладнення і обгрунтовує проведення корегуючої терапії.
Показник загального Са крові знижувався на тлі зменшення мінеральної щільності кісткової тканини (МЩКТ). Протилежні зміни відбувалися з Са сечі: найменші вони були у пацієнтів з нормальним станом МЩКТ і найбільшими – у хворих з остеопорозом. Виявлено кореляційну залежність між рівнем загального Са сироватки крові та сечі, а також показниками МЩКТ.
Концентрація іонів Са в сироватці крові є однією з найбільш жорстких констант: гіпокальціємія завжди стимулює лінійне підвищення рівня паратиреоїдного гормону, ефект дії якого досягається шляхом мобілізації Са з кісток у позаклітинну рідину. Цей постулат підтверджується тим, що зменшення вмісту загального Са крові в межах 1 % приводить до його виходу з депо – кісткової тканини. Зниження рівня Са в сироватці крові з подальшим розвитком вторинного гіперпаратиреозу індукує процес остеопатії, призводить до таких ускладнень, як остеомаляція, зменшення мінеральної щільності кісткової тканини і остеопорозу.
Наявність у складі жовчі солей Са і таких стимуляторів його всмоктування в кишечнику як вітамін D і жовчні кислоти робить жовчоутворення і жовчовиділення важливим чинником у механізмах регуляції кальцієвого гомеостазу. Тому при холестатичних захворюваннях печінки також спостерігається розвиток вторинного остеопорозу.
Прогресуючий спадковий внутрішньопечінковий холестаз являє собою гетерогенну групу аутосомно-рецесивних захворювань печінки, що характеризуються раннім розвитком остеопорозу та дебютом холестазу, який прогресує перед настанням пубертатного періоду, перетворюючись у цироз і печінкову недостатність. Існує три типи прогресуючого спадкового внутрішньопечінкового холестазу:
Перший тип викликається дефектами біосинтезу жовчних кислот і найчастіше проявляється в ранньому дитинстві.
Другий тип, хвороба Байлера, є результатом недостатньої секреції солей жовчних кислот. Симптоми захворювання починаються пізніше у вигляді приступів жовтяниці, сверблячки й кон’югованої гіпербілірубінемії, що часто провокуються інтеркурентними захворюваннями. Незважаючи на те, що пацієнти спочатку повністю видужують у проміжку між приступами, функція печінки повільно погіршується та до віку 15 років за звичай наступає важка печінкова недостатність. Оптимальним шляхом лікування стає пересадження печінки, проведення якого може ускладнюватися через серйозні енцефалопатичні реакції. Ідентифіковано два локуси хвороби Байлера, один із яких локалізований на 18-й хромосомі (PFІ1), а іншої на – 2-й хромосомі (PFІ2).
Висока активність γ-глутамілтрансферази у сироватці крові та зміни в гістологічній картині печінки, що проявляються запаленням ворітної вени та проліферацією канальців у ранній стадії розвитку захворювання, відрізняють третій тип прогресуючого спадкового внутрішньо-печінкового холестазу від двох перших типів, що викликані дефектами синтезу жовчних кислот або дефектами секреції.
Патологія кісток є ускладненням більшості спадкових захворювань печінки, що супроводжуються холестазом. До цієї групи можна віднести пігментні гепатози. Болі в кістках і переломи при таких станах, що спостерігаються у деяких пацієнтів обумовлені розвитком остеомаляції та остеопорозу.
Синдром Жильбера являє собою доброякісну сімейну некон’юговану помірну гіпербілірубінемію (рівень білірубіну в сироватці в межах 17-85 мкмоль/л), не пов'язану з гемолізом і яка має доброякісний перебіг. Гіпербілірубінемія носить сімейний характер і не супроводжується порушенням біохімічних показників функції печінки та її гістологічної картини. У популяції частота синдрому Жильбера становить 2-5 %. Він може бути випадково виявлений при профілактичному медичному обстеженні або при обстеженні з приводу іншого захворювання (наприклад, вірусного гепатиту). Прогноз сприятливий. Жовтяниця виражена помірно й має інтермітуючий характер. Вона може підсилюватися після інтеркурентних інфекцій або після голодування, іноді супроводжується слабкістю, нудотою і часто неприємними відчуттями в області печінки. Ці симптоми, можливо, виражені не більш ніж у здорових людей без гіпербілірубінемії. Яких-небудь інших симптомів при обстеженні зазвичай виявити не вдається, селезінка не пальпується.
При синдромі Жильбера знижується зв'язування білірубіну із глюкуроновою кислотою в печінці до 30 % від нормального. В основі захворювання лежить генетичний дефект – наявність на промоторній ділянці (А(ТА)6ТАА) гена, що кодує УДФГТ 1*1, додаткового динуклеотиду ТА, що призводить до утворення ділянки (А(ТА)7ТАА). Цей дефект успадковується за аутосомно-рецесивном типом, тому для розвитку недуги хворий повинен бути гомозиготою за цим алелем. Вважають, що подовження промоторної послідовності порушує зв'язування фактору транскрипції ІІD, що приводить до зменшення утворення ферменту УДФГТ 1. Однак одного тільки зниження синтезу ферментів для розвитку синдрому Жильбера недостатньо; необхідно також наявність інших факторів, наприклад, схованого гемолізу та порушення транспорту білірубіну в печінці.
У клітинах периферичної крові виявляються порушення, що нагадують змішану порфірію, імовірно внаслідок збільшення концентрації білірубіну в клітинах печінки.
Спеціальні діагностичні проби при синдромі Жильбера включають пробу з голодуванням (підвищення рівня білірубіна в сироватці на тлі голодування), пробу з фенобарбіталом (прийом фенобарбітала, що індукує кон’югуючі ферменти печінки, викликає зниження рівня білірубіна), пробу з нікотиновою кислотою (внутрішньовенне введення нікотинової кислоти, що зменшує осмотичну резистентність еритроцитів, викликає підвищення рівня білірубіна). Тонкошарова хроматографія виявляє значно більш високу частку (у порівнянні з нормою) некон’югованого білірубіну при хронічному гемолізі або хронічному гепатиті, що має діагностичне значення. При біопсії печінки виявляють зниження вмісту кон’югуючих ферментів. Однак синдром Жильбера зазвичай вдається діагностувати, не вдаючись до цих спеціальних методів дослідження.
Тривалість життя при синдромі Жильбера не нижча, ніж у здорових людей, тому спеціального лікування не потрібно і хворого досить лише заспокоїти. Гіпербілірубінемія зберігається постійно, однак підвищення смертності не відбувається. Рівень білірубіна в сироватці можна знизити за допомогою фенобарбіталу, але оскільки жовтяниця зазвичай виражена незначно, косметичний ефект від такого лікування відзначається лише у деяких хворих. Необхідно попереджати хворих, що жовтяниця може з'явитися після інтеркурентних інфекцій, повторної блювоти і пропущеного прийому їжі.
Синдром Криглера-Найяра – форма сімейної негемолітичної жовтяниці, при якій рівень некон'югованного білірубіна в сироватці дуже високий. У печінці можна виявити недостатність кон'югуючого ферменту. Концентрація пігменту в жовчі мінімальна. Незважаючи на токсичні прояви гіпербілірубінемії, бромсульфалеїнова проба негативна.
Синдром Криглера-Найяра типу 1 успадковується за аутосомно- рецесивним типом. У печінці відсутня активність кон’югуючого ферменту, у жовчі – кон’югований білірубін. У сироватці не виявляються глюкуроніди білірубіну. Оскільки рівень білірубіна в сироватці згодом стабілізується, варто припустити існування альтернативного шляху метаболізму білірубіна [169].
На молекулярному рівні дефект локалізується в одному з п'яти екзонів (1А-5) гена УДФГТ 1*1.
Зазвичай, хоча й не завжди, хворі гинуть протягом першого року життя на тлі ядерної жовтяниці. Прийом фенобарбіталу неефективний. Кровопускання й плазмаферез, що застосовувалися для зниження рівня білірубіна в сироватці, давали лише тимчасовий ефект. При фототерапії рівень білірубіна в сироватці вдається знизити майже на 50 %; цей метод лікування можна застосовувати амбулаторно. Протягом першого або другого десятиріччя життя в будь-який момент може розвитися енцефалопатія (ядерна жовтяниця). Після трансплантації печінки обмін білірубіну нормалізується, гіпербілірубінемія зникає, прогноз поліпшується . Трансплантацію варто проводити в молодому віці, особливо якщо проведення фототерапії неможливе.
Синдром Криглера-Найяра типу 2 також успадковується за аутосомно- рецесивним типом. Активність ферменту, що здійснює кон'югацію білірубіна в печінці, значно знижується (до 10 % від нормальної й менш) і не визначається звичайними методами. Призначення фенобарбіталу приводить до вираженого ефекту, і хворі доживають до дорослого віку.
При аналізі ДНК гена УДФГТ 1*1 виявлені мутації в екзонах 1А-5. Проте при вивченні експресії цих мутантних генів виявлена залишкова активність ферменту, і цим пояснюються менш висока, ніж при синдромі Криглера- Найяра типу І, білірубінемія, наявність глюкуронидів у сечі й ефективність лікування фенобарбіталом.
У деяких родичів хворих із синдромом Криглера-Найяра виявляється підвищення рівня білірубіна в сироватці, що хоча й не настільки високе, як у хворих, але перевищує його рівень при синдромі Жильбера. Аналіз гена УДФГТ 1*1 дозволяє припустити, що у таких пацієнтів є змішана гетерозиготність: в одному з аллелей є мутація ТАТАА, властива синдрому Жильбера, а в іншому – мутація, властива синдрому Криглера-Найяра.
Синдром Криглера-Найяра типу 2 не завжди протікає доброякісно, і для забезпечення рівня білірубіна нижче 450 мкмоль/л потрібне поєднання фототерапії із призначенням фенобарбіталу.
Розрізнити типи 1 і 2 синдрома Криглера-Найяра можна оцінивши ефективність лікування фенобарбіталом шляхом визначення фракцій білірубіну за допомогою високоефективної рідинної хроматографії або визначаючи зміст у жовчі жовчних пігментів після призначення фенобарбіталу. При типі 2 рівень білірубіна в сироватці частка некон’югованого білірубіна знижуються, а вміст моно- і дикон’югатів у жовчі збільшується. При типі 1 рівень білірубіна в сироватці не знижується, а в жовчі виявляється переважно некон’югований білірубін. Очевидно, у майбутньому діагностика буде ґрунтуватися на експресії іn vіtro мутантної ДНК хворих. Пацієнти із синдромом Криглера-Найяра, безсумнівно, є кандидатами на генотерапію.
Синдром Дубіна-Джонсона – хронічне доброякісне захворювання, що проявляється непостійною жовтяницею з підвищенням рівня переважно кон’югованого білірубіна та білірубінурією. Він успадковується за аутосомно- рецесивним типом, розповсюджений переважно на Середньому Сході серед іранських євреїв. В основі синдрому лежить погіршення транспорту в жовч багатьох органічних аніонів, які не відносяться до жовчних кислот, що обумовлено дефектом АТФ-залежної транспортної системи канальців.
Макроскопічно печінка має зеленувато-чорний колір (жовтяниця із чорною печінкою). При гістологічному дослідженні в гепатоцитах виявляється коричневий пігмент, що не є ні залізом, ні компонентом жовчі. Кількість цього пігменту в печінці та рівень білірубінемії не зв'язані між собою. Хімічний склад пігменту не вивчений. Раніше вважали, що це меланін, але наявні в цей час дані свідчать про те, що він утворюється в результаті порушення секреції аніонних метаболітів тирозину, фенілаланіну й триптофану. Синдром Дубіна-Джонсона не супроводжується сверблячкою; активність лужної фосфатази й рівень жовчних кислот у сироватці зберігаються в межах нормальних значень. Екскреція органічних аніонів у жовч порушується. Їхнє поглинання печінкою не страждає. Вміст копропорфіринів у сечі нормальний, але частка ізомеру типу I збільшується. Захворювання може вперше виявитися жовтяницею при вагітності або на тлі прийому пероральних контрацептивів (ці стани викликають погіршення екскреторної функції печінки). Прогноз сприятливий.
Синдром Ротора – форма хронічної сімейної гіпербілірубінемії з підвищенням некон’югованої фракції білірубіну нагадує синдром Дубіна- Джонсона. Основною відмінністю її від останнього служить відсутність коричневого пігменту в гепатоцитах. Крім того, при синдромі Ротора жовчний міхур при холецистографії контрастується, а при бромсульфалеїновій пробі –вторинного підвищення концентрації барвника не відбувається. Загальний рівень копропорфіринів у сечі підвищений, як при холестазі. Ізомер типу І копропорфірину в сечі становить приблизно 65 % всіх копропорфіринів. При електронній мікроскопії можуть виявлятися патологічні зміни мітохондрій і пероксисом. Вивчення сімейного анамнезу дозволяє припустити можливість аутосомного успадкування. Прогноз сприятливий.
Холестаз може бути результатом синдрому Алажиля, також відомого як артеріопечінкова дисплазія (синдром збідніння жовчних проток), розповсюдженого в усьому світі. Ознаки хронічного внутрішньопечінкого холестаза виникають у немовлят або дітей раннього віку. Захворювання успадковується за аутосомно-домінантним типом із різною експресією та пенетрантністю. Виявлена делеція короткого плеча в хромосомі 20. У фенотипі пацієнтів звертає на себе увагу обличчя трикутної форми із широким опуклим чолом, глибоко посадженими очима, сплющеним носом і гострим підборіддям. Зазвичай виявляють гепатоспленомегалію. Зміни скелету включають укорочені дистальні фаланги й деформацію тіл хребців у формі метелика. Часто виявляють аномалії розвитку нирок і стеноз периферичних гілок легеневої артерії.
При біопсії печінки виявляється, що кількість міждолькових жовчних протоків зменшена, вони також можуть бути відсутніми, зменшена кількість портальних зон. У зв'язку з невеликою виразністю фіброзу цироз печінки й портальна гіпертензія не розвиваються. Однак для діагностики синдрому Алажиля дані біопсії печінки не є вирішальними.
Хворі доживають до дорослого віку, при цьому в них спостерігається різна ступінь розумової відсталості, відставання в рості, прояви ксантоматозу й шкірної сверблячки. Може розвиватися гепатоцеллюлярна карцинома, унаслідок чого виникає печінкова остеодистрофія.
У дослідженнях Hopf U. (1998) зі співав. було встановлено зменшення проліферації остеобластів під дією плазми, отриманої від хворих із жовтяницею; при цьому інгібіруючий ефект надавав некон’югований білірубін, а не жовчні кислоти. Ці данні дозволяють пояснити порушення формування кісткової тканини при хронічному спадковому холестазі, однак вони потребують подальших досліджень.
Рентгенологічно у хворих із проявами хронічного холестазу виявляють зміни, характерні для остеомаляції: псевдопереломи, зони Лоозера. Рентгенографія кистей виявляє розрідження кісткової тканини. При біопсії кісток виявляються широкі некальціфіковані остеоїдні маси, що оточують трабекули.
При дослідженні кальцієвого обміну у пацієнтів із хронічними холециститами (Власенко О.В., 2004) визначався вміст загального та іонізованого Са в крові, жовчі та сечі. Також визначався показник відносного вмісту в крові іонізованого Са до загального, який відображає розподіл даного мікроелементу між біологічними структурами – клітиною і міжклітинним простором.
При зборі анамнезу у хворих було звернуто увагу на той факт, що майже у 20 % із них були посилання на перенесені раніше переломи при мінімальній травмі (компресійні переломи тіл хребців, дистальних відділів променевої кістки, проксимальних відділів плечової і стегнової кісток), епізоди гострого болю в хребті, що передували перелому за кілька років.
При загостренні холециститу близько 17 % обстежених вказували на появу гострих або загострення хронічних болів у хребті, що посилюються при русі; близько 40 % пацієнтів відзначали біль у литкових м'язах або великих суглобах. Аналіз кальцієвого балансу в сироватці крові показав зниження загального (1,2 разів) і іонізованого (1,25 рази) Са у пацієнтів основної групи. Незважаючи на здавалося б однакові цифрові зміни в даних показниках, співвідношення іонізованого Са до загального склало 47,2 % при нормі 46,1 %. Тобто порушується біопотенціал клітини, тому що збільшення показника відношення іонізованого Са до загального вказує на значне порушення екстра- та інтрацеллюлярного розподілу Са²+, відображає вираженість процесів нервово-м'язового збудження, будучи, мабуть, одним із патогенетичних механізмів діскінетичних розладів жовчного міхура (ЖМ) та сфінктера Одді.
Порівняння показників кальцієвого балансу в залежності від типу дискінезії показало, що найбільші зміни в кальцієвому обміні відзначені у хворих з дискінезією ЖМ за гіпомоторним типом, чому, на нашу думку, сприяв не тільки застій жовчі в міхурі, але і зменшення її надходження в кишечник у результаті порушень тонусу сфінктера Одді.
При дослідженні вмісту загального Са жовчі відзначено його збільшення в 4,7 рази в порівнянні зі здоровими особами. Зіставлення вмісту Са в жовчі з показниками холато-холестеринового коефіцієнту (ХХК) виявило зворотну залежність, яка була більш вираженою у хворих із гіпомоторним типом дискінезії ЖМ (r =- 0,72). Така зміна фізико-хімічних властивостей жовчі розглядається як індикатор схильності до каменеутворення і формування хронічного калькульозного холециститу.
Гомеостаз Са і тут відіграє дуже важливу роль. Експериментальними роботами було доведено, що іони Са різко збільшують активність тканинних фосфатаз і лужної фосфатази. Це призводить до зростання секреції жовчі, концентрації в ній ліпідних сполук, жовчних кислот і посилення секреції білірубіну. Зв'язування вільних іонів Са²+ в жовчі з жовчними кислотами є важливим чинником, який стає перешкодою для утворення важкорозчинних кальцієвого осаду, тобто важкорозчинні солі Са можуть утворюватися тільки в умовах порушення біосинтезу жовчних кислот та інших органічних сполук.
Зважаючи на вищевказані механізми, можна зробити висновок, що дісметаболізм Са є найважливішим чинником порушення не тільки кінетики жовчовивідних шляхів (ЖВШ), а й самого процесу секреції жовчі та її складу. В умовах гіпокальціємії змінюються кількісні та якісні характеристики жовчоутворення, зростає коефіцієнт літогенність жовчі і порушуються процеси її виведення, особливо на тлі запалення ЖМ. Тобто наявність гіпокальціємії є одним із найважливіших факторів, який сприяє каменеутворенню в ЖМ.
З іншого боку, саме важкість перебігу хронічного некалькульозного холециститу (ХНХ) та вираженість біліарної дисфункції є факторами ризику розвитку гіпокальціємії. Зіставлення показників кальцію крові з типом дискінезії жовчного міхура (ДЖМ), вираженістю больового синдрому, наявністю збудника в жовчі і тривалістю захворювання показало, що найбільші зміни в гомеостазі Са відзначені у хворих на ХНХ з тривалістю анамнезу більше 10 років, частими рецидивами захворювання, наявністю вираженої біліарної дисфункції (гіпомоторний тип ДЖМ та дистонія сфінктера Одді). При цьому вираженість больового синдрому перебуває в пропорційній залежності від співвідношення іонізованого до загального Са крові.
У дослідженнях Софронової О.М. (2006 р.) було доведено, що на величини показників кальцієвого гомеостазу негативний вплив мають також супутня патологія: підвищений артеріальний тиск і підвищена маса тіла (остання супроводжувалася розвитком патології печінки – стеатогепатоза або стеатогепатиту). А поєднання післяхолецистектомічного синдрому (ПХЕС) і гіпертонічної хвороби (ГХ) призводило до формування метаболічного синдрому, перебіг якого був обтяжений розвитком змін кальцієвого обміну. Так, рівень загального та іонізованого Са в крові у хворих з ПХЕС і ГХ були знижені (р<0,01) і мали залежність від тривалості анамнезу як жовчнокам'яної хвороби та ПХЕС, так і артеріальної гіпертензії (максимальне зниження відзначено при тривалості анамнезу більше 10 років), стадії ГХ, наявності інсулінорезистентності, величини індексу маси тіла (ІМТ).
Для визначення вираженості остеопенічного змін проводилися денситометричної дослідження структури кісткової тканини у даних хворих (Пасієшвілі Л.М., Власенко Е.В., 2004). Так, при обстеженні 37 пацієнтів із високим ризиком зниження мінеральної щільності кісток (переломи кісток в анамнезі, середній ступінь тяжкості екскреторної недостатності ПЗ, інсулінорезистентність, менапаузальний синдром) частота остеопорозу у жінок у віці 40-49 років склала 28,9 %, 50 років і старше серед жінок – 41,2 %, серед чоловіків – 29,8 %. Для порівняння в московській популяції («практично здорові особи») частота остеопорозу у віці 50 років і старше серед жінок склала 33,8 %, а у чоловіків – 26,9 % (за даними денситометрії поперекового відділу хребта і проксимального відділу стегна) (Беневоленская Л.И., 2003). В Україні, згідно з даними В.В. Поворознюка (2000 р.), у жінок у віковій групі 50-59 років ознаки остеопорозу визначалися у 13 % випадків, в 60-69 років – у 25 %, в 70-79 років – у 50 % і в 80-89 років – у 53 %.
Оригінальні дослідження дозволили розробити і запропонувати для застосування у практичній охороні здоров'я алгоритм обстеження хворих з патологією біліарної системи для діагностики остеопорозу та профілактики переломів (Бобро Л.Н., Пасиешвили Л.М., 2008).
Таким чином, проведені власні дослідження, а також аналіз даних літератури дозволяють говорити про те, що більшість спадкових і набутих захворювань ШКТ є Са-залежними. Їх хронічний рецидивуючий перебіг супроводжується порушенням кальцієвого обміну різного ступеня, що призводить до формування остеопенічного стану та остеопорозу.
Зниження вмісту Са в крові і особливо перерозподіл його між біологічними рідинами визначає клінічну симптоматику основного захворювання, яке призвело до розвитку гіпокальціємії, ступінь проявів больового та диспепсичного синдромів, що необхідно враховувати при проведенні терапії та реабілітації пацієнтів із захворюваннями органів травлення.

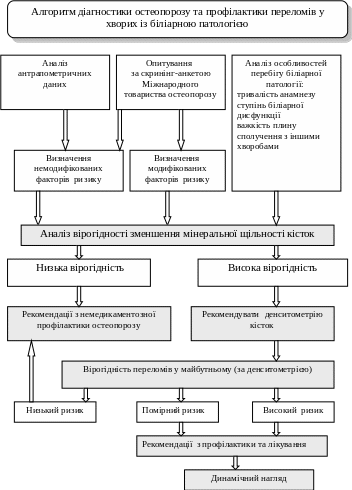
Рис. 1. Алгоритм обстеження хворих з патологією біліарної системи для діагностики остеопорозу та профілактики переломів
(за даними Бобро Л.Н., Пасиешвили Л.М., 2008)