

Основы_Генетики_1А
.pdfРисунок V, 46. Ребенок с синдромом кошачьего крика. По материалам сайта http://medarticle23.moslek.ru/articles/34943.htm.
Синдром Вольфа-Хиршхорна (синдром 4p) возникает по причине дефишенси (терминальной делеции) короткого плеча хромосомы 4. Синдром встречается с частотой 1 на 96000 живых новорожденных. У большинства больных делетирован район 4p16.3, величина делеции – около 165 т.п.н. При меньшем размере делетированного участка проявляется менее выраженный фенотип. Использование рутинной окраски хромосом позволяет выявить эту аберрацию только в 60% случаев. Флуоресцентная гибридизация in situ дает возможность выявлять до 95% носителей. Для больных характерна микроцефалия, задержка внутриутробного развития, «рыбий» рот, краниофациальный дисморфизм, расщелину губы и
131

неба, низкорасположенные деформированные ушные раковины, гипертелоризм (Рисунок V, 47). Из внутренних органов чаще всего поражаются почки (диплазия, кисты), сердце (атриальный или вентрикулярный септальные дефекты) и семенники (крипторхизм). Выживает не более 20% новорожденных с этим синдромом, продолжительность жизни – до 25 лет. У больных наблюдается грубая задержка умственного развития.
Рисунок V, 47. Ребенок с синдромом Вольфа-Хиршхорна. По материалам сайта http://en.wikipedia.orgwikiWolf-Hirschhorn_syndrome
В некоторых популяциях человека широко распространены перицентрические инверсии inv 9 (p11; q13) (Рисунок V, 48) и inv 10 (p11; q21) (Рисунок V, 49), не имеющие фенотипического проявления. Хотя некоторые авторы указывают на возможность снижения фертильности у их носителей, поскольку инверсии затрудняют
132

коньюгацию гомологичных хромосом у гетерозигот и подавляют кроссинговер на своих участках.
Рисунок V, 48. Перицентрическая инверсия HSA9. Фотография Е.Г. Нероновой.
133
Рисунок V, 49. Перицентрическая инверсия HSA10. Фотография Е.Г. Нероновой.
В популяциях человека часто встречается полиморфизм (биологическое разнообразие) длины гетерохроматиновых районов, в том числе вторичных перетяжек. Такие изменения принято обозначать h+ - увеличение гетерохроматинового района (в том числе вторичных перетяжек) по сравнению с нормой и h- - уменьшение. Как правило, фенотипического эффекта такие полиморфные варианты не имеют.
Примеры:
46, 15 ph+ - увеличение длины вторичной перетяжки в коротком плече хромосомы 15;
46, 21ph- - уменьшение длины вторичной перетяжки в коротком плече хромосомы 21;
46, XY, Yqh+ - увеличение гетерохроматинового района Y- хромосомы.
Причиной хронического миелобластного лейкоза (ХМЛ)
является соматическая мутация - слияние части гена тирозинкиназы ABL1 9-й хромосомы с геном BCR 22-й хромосомы с образованием химерного белка (Рисунок V, 50) . Филадельфийская хромосома, выявляемая у всех страдающих ХМЛ, - результат реципрокной транслокации t (9; 22), (q34; q11). Название «филадельфийская» этой аберрантной хромосоме дано потому, что впервые ее обнаружили
134

сотрудники Пенсильванского университета в Филадельфии. ХМЛ - миелопролиферативное заболевание, при котором в норме находящиеся в фазе G0 гранулоциты (нейтрофилы, базофилы и эозинофилы) начинают активно делиться под влиянием химерного белка BCR/ABL1. В хронической фазе заболевания больные обычно чувствуют недомогание и переполнение живота. Акселеративная фаза заболевания может привести к бластному кризу, который протекает подобно острому лейкозу с быстрой прогрессией и небольшой выживаемостью.
Рисунок V, 50. Филадельфийская хромосома.
135
Синдром Дауна иногда имеет транслокационную природу. Значительная часть хромосомы 21 может быть перемещена на другие хромосомы (чаще на 15, реже на 14, ещё реже на 21, 22 и Y- хромосому). В таких случаях при нормальном числе хромосом функционально будет присутствовать анеуплоидия по хромосоме 21.
На хромосоме X с частотой 1 на 1000 – 1 на 2000 новорожденных мальчиков в районе q27-28 наблюдается вторичная перетяжка – так называемый фрагильный сайт. Это синдром Мартина-Белл или синдром ломкой X-хромосомы. Молекулярный механизм заключается в экспансии тринуклеотидных ЦГГ (цитозин- гуанин-гуанин) повторов. В норме в районе Xq27.3 должно быть от 6 до 54 таких повторов, предмутационное состояние – от 55 до 200 таких повторов, этом случае у матерей с предмутационным состоянием возможно увеличение числа повторов в мейозе из-за неравного кроссинговера, и их потомки могут получить мутантную (с числом повторов более 200) X-хромосому. Наиболее выражено появление синдрома в гемизиготе, то есть у мальчиков с мутантной по фрагильному сайту X-хромосомой. Такие мальчики рождаются с весом 3,5 – 4 кг и макроорхизмом. Часто наблюдаются увеличенные размеры головы, длинное лицо с увеличенным подбородком, низкое расположение ушных раковин, повышенная подвижность суставов (Рисунок V, 51). Главные симптоматические признаки – умственная отсталость и своеобразные нарушения речи – эхолалия (неосмысленное повторение чужих слов) и персеверация (бормочущая речь). Иногда отмечается ранний детский аутизм.
136

Рисунок V, 52. Синдром Мартина-Белл. По материалам сайта http://blog.ahfr.org/2008/05/fragile-x-syndrome.html
Следует отметить, что для всех хромосомных аберраций характерны общие фенотипические проявления: низкий вес при рождении, задержка развития, низкий рост, микроцефалия, микрогнатия, нарушения остеогенеза, аномальное расположение глаз.
Контрольные вопросы и задания к главе V
1. Прочитайте кариологический диагноз:
а) 46, XX, t (1; 22), (q24; q12)
б) 46, XX, inv 7 (p11; p13)
137

в) 47, XY, +22, r (5)
г) 48, XXYY
д) 46, XX, inv 4 (q11; q21)
е) 45, X, inv X (p11; q13)
ж) 46, XY, i (4q)
з) 48, XX, +21, +16p+
и) 45, X, 22ph+
2. Поставьте кариологический диагноз на основе фотографий митотических хромосом на рисунке V, 52.
А.
138
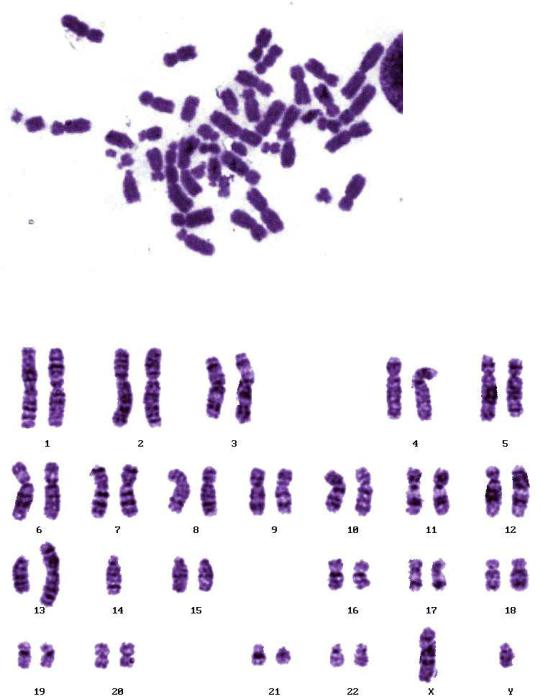
Б.
В.
139

Г.
Д.
140