

Основы_Генетики_2
.pdfчто определяет разную степень выраженности симптомов заболевания. Например, галактоземия Дурте протекает почти бессимптомно, отмечается только склонность к расстройствам печени.
Болезнь Гирке (гликогеноз I типа, гликогеновая болезнь I типа) –
неспособность превращения глюкозо-6-фосфата в глюкозу, которая приводит к нарушению синтеза и разложения гликогена.
Депонирование гликогена происходит, обратный процесс – нет.
Развивается гипогликемия. Накопление избыточного количества гликогена в печени и почках приводит к печеночной и почечной недостаточности. Тип наследования – аутосомно-рецессивный.
Причина заболевания – мутация в гене G6PC , который кодирует фермент глюкозо-6-фосфатазу. Описано 14 мутантных аллелей этого гена, которые связаны с болезнью Гирке. Существуют молекулярно-
генетические тесты для выявления гетерозиготного носительства и пренатальной диагностики этого заболевания.
3. Нарушения липидного обмена
Болезнь Ниманна-Пика типов А и Б - снижение активности фермента кислой лизосомальной сфингомиелиназы, который кодируется геном
SMPD1 (HSA11p15.4-p15.1). Тип наследования – аутосомно-
рецессивный. Нарушение липидного метаболизма приводит к накоплению липидов в печени, легких, селезенке, нервных тканях.
Характерна дегенерация нервных клеток, нарушение деятельности нервной системы, повышенный уровень холестерина и липидов в крови. Тип А летален в раннем детском возрасте. Тип Б протекает
233
более мягко, больные как правило доживают до взрослого состояния.
Разные типы обусловлены разными мутациями в гене SMPD1.
Болезнь Гоше (гликозилцерамидный липидоз) - накопление глюкоцереброзидов в клетках нервной и ретикуло-эндотелиальной системы, обусловленное дефицитом фермента глюкоцереброзидазы,
которая кодируется геном GBA (HSA1q21). Относится к группе лизосомных болезней накопления. Некоторые формы заболевания проявляются в тяжелых поражениях печени, селезенки, нервной и костной тканей.
4. Наследственные болезни пуринового и пиримидинового обмена
Синдром Леша-Нихена – сцепленное с полом рецессивное заболевание, при котором резко возрастает содержание мочевой кислоты во всех жидкостях тела. Последствием этого является задержка развития, умеренная умственная отсталость, приступы агрессивного поведения с самоповреждением. Недостаточность ферментативной активности гипоксантин-
гуанинфосфорибозилтрансферазы по причине мутаций в гене HPRT1
(HSAXq26-q27.2) лежит в основе этого заболевания. Описаны несколько мутаций в том же гене, следствием которых является
подагра (нарушение пуринового обмена и отложение мочекислых соединений в тканях).
5. Нарушения обмена соединительной ткани
Синдром Марфана («паучьи пальцы», арахнодактилия) - поражение соединительной ткани вследствие мутации в гене FBN1 (HSA15q21.1),
234

ответственном за синтез фибриллина. Наследуется по аутосомно-
доминантному типу. Клиническая полиморфность заболевания объясняется большим числом мутантных аллелей, каждый из которых может проявляться в гетерозиготном состоянии. Для больных характерен высокий рост, астеническое телосложение
(непропорционально длинные конечности), арахнодактилия (длинные тонкие пальцы), слабость связочного аппарата, отслойка сетчатки глаза, подвывих хрусталика, пролапс митрального клапана (Рисунок
IX, 2).
Рисунок IX, 2. Синдром Марфана. По материалам сайта http://www.spineinfo.ru/infosources/case/cases_14.html.
235
Мукополисахаридозы - группа заболеваний соединительной ткани,
связанных с нарушеним обмена кислых гликозаминогликанов
(мукополисахаридов), вызванных недостаточностью некоторых лизосомных ферментов. Эти заболевания относят к лизосомным болезням накопления. Они проявляются в различных дефектах костной и соединительной тканей. Мукополисазаридоз типа I (синдром Хурлера) – аутосомно-рецессивное заболевание,
возникающее в результате дефицита фермента альфа-L-идуронидазы из-за мутаций в гене IDUA (HSA4q16.3). Это приводит к накоплению белково-углеводных комплексов и жиров в клетках организма. В
результате у больных наблюдается малый рост, существенная задержка умственного развития, увеличение печени и селезенки,
пороки сердца, помутнение роговицы, деформация костей и огрубение черт лица (Рисунок IX, 3).
236

Рисунок IX, 3. Синдром Хурлера. По материалам сайта http://medgen.genetics.utah.edu/photographs/pages/hurler_syndrome.htm.
Мукополисахаридоз типа II (синдром Хантера) – сцепленное с полом рецессивное заболевание, которое обусловлено дефектом фермента идуронатсульфотазы из-за мутации в гене IDS (HSAXq28).
Веществами накопления являются дерматан- и гепарансульфаты.
Характерны грубые черты лица, скафоцефалия, шумное дыхание,
низкий грубый голос, частые острые респираторные вирусные инфекции (Рисунок IX, 4). В возрасте 3—4 лет появляются нарушения координации движений — походка становится неуклюжей, дети при ходьбе часто падают. Для больных характерны эмоциональная лабильность и агрессивность. Наблюдаются также прогрессирующая
237
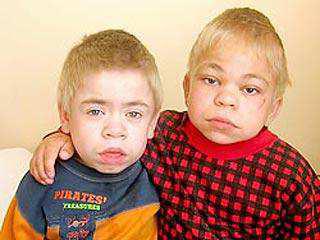
тугоухость, узелковые поражения кожи спины, остеоартриты,
поражения роговицы.
\
Рисунок IX, 4. Синдром Хантера. По материалам сайта
http://1nsk.ru/news/russia/23335.html.
Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо)- заболевание, вызванное накоплением гепарансульфата.
Для него характерна генетическая гетерогенность – существуют 4
типа этой болезни, вызванные мутациями в 4-х разных генах,
кодирующих ферменты, участвующие в метаболизме накапливаемого вещества. Первые симптомы болезни в виде нарушений сна появляются у детей старше 3 лет. Постепенно развивается апатия,
отмечается задержка психомоторного развития, нарушения речи,
черты лица становятся грубыми. Со временем дети перестают узнавать окружающих. Для больных арактерны задержка роста,
контрактуры суставов, гипертрихоз, умеренная гепатоспленомегалия.
В отличие от синдромов Хурлера и Хантера при болезни Санфилиппо преобладает умственная отсталость, а поражения роговицы и сердечно-сосудистой системы отсутствуют.
238

Рисунок IX, 5. Синдром Санфилиппо. По материалам сайта
http://runkle-science.wikispaces.com/Sanfilippo-syndrome.
Фибродисплазия (оссифицирующий миозит, параоссальная
гетеротопическая оссификация, болезнь Мюнхеймера) - заболевание соединительной ткани, связанное с ее прогрессирующим окостенением в результате мутации в гене ACVR1 (HSA2q23-q24),
который кодирует рецептор активина А. Тип наследования – аутосомно-доминантный. Заболевание проявляется врожденными дефектами развития — прежде всего искривленными большими пальцами стоп и нарушениями в шейном отделе позвоночника на уровне позвонков с2 — с7. Заболевание имеет прогредиентный характер, приводит к значительным нарушениям функционального
239

состояния опорно-двигательного аппарата, глубокой инвалидизации больных и смерти преимущественно в детском и молодом возрасте
(Рисунок IX, 6). Болезнь еще называют «болезнь второго скелета», так как там где в организме должны происходить штатные противовоспалительные процессы, начинается рост кости.
Рисунок IX, 6. Фибродисплазия. По материалам сайта http://donbass.ua/news/health/2010/02/15.
6. Нарушения циркулирующих белков
Гемоглобинопатии - наследственные нарушения синтеза гемоглобина.
Различают две группы гемоглобинопатий. Для первой характерно изменение первичной структуры белка глобина, что может сопровождаться нарушениями его стабильности и функции
(например, серповидноклеточная анемия). При гемоглобинопатиях
240
второй группы структура гемоглобина остается нормальной, снижена лишь скорость синтеза глобиновых цепей (например, β-талассемия).
7. Нарушения обмена веществ в эритроцитах
Наследственный сфероцитоз - врождённая недостаточность липидов оболочки эритроцитов. Для заболевания характерен аутосомно-
доминантный или аутосомно-рецессивный тип наследования в зависимости от мутации гена SPTA1 (HSA1q21), который кодирует эритроцитарный α-1 спектрин. Аномалия этого белка приводит к повышению концентрации ионов натрия внутри эритроцита, и
проникновению в него избытка воды из-за повышения осмотического давления. Вследствие этого образуются сферические эритроциты – сфероциты, которые в отличие от двояковогнутых нормальных эритроцитов, не обладают способностью изменять форму в узких участках кровотока, например при переходе в синусы селезенки. Это приводит к замедлению продвижения эритроцитов в синусах селезенки и отщеплению части мембраны эритроцита с образованием микросфероцитов. Разрушенные эритроциты поглощаются макрофагами селезенки. Гемолиз эритроцитов приводит к гиперплазии клеток пульпы и увеличению селезенки. Одним из основных клинических симптомов является желтуха. Основными симптомами наследственного сфероцитоза являются увеличение селезенки (обычно выступает из-под подреберья на 2 — 3 см) и
желтуха. Иногда наблюдаются признаки замедленного развития,
нарушения лицевого скелета, башенный череп, седловидного носа,
высокого стояния неба, нарушения расположения зубов, узких глазниц.
241
8. Наследственные болезни обмена металлов
Болезнь Коновалова-Вильсона (гепатоцеребральная дистрофия) –
аутосомно-рецессивное нарушение метаболизма меди, приводящее к тяжелейшим поражениям центральной нервной системы и внутренних органов. Заболевание обусловлено низким или аномальным синтезом церулоплазмина (белка, транспортирующего медь) из-за недостаточности ферментативной активности медь-переносящей АТФазы. Мутации (их описано около 200) в гене ATP7B (HSA13q14q21) приводят к изменениям β-полипептида этого фермента, что является генетической основой этой патологии. Основную роль в патогенезе играет нарушение обмена меди, её накопление в нервной,
почечной, печёночной тканях и роговице, вследствие чего происходит токсическое повреждение медью данных органов. В печени формируется крупноузловой или смешанный цирроз. В почках в первую очередь страдают проксимальные канальцы. В головном мозге поражаются в большей степени базальные ганглии, зубчатое ядро мозжечка и черная субстанция.
9. Нарушения всасывания в пищеварительном тракте
Муковисцидоз (кистозный фиброз) — аутосомно-рецессивное заболевание, характеризующееся поражением желез внешней секреции, тяжёлыми нарушениями функций органов дыхания и желудочно-кишечного тракта. Причиной являются мутации гена
CFTR (HSA7q31.2), который кодирует трансмембранный регулятор кистозного фиброза. Заболевание характеризуется поражением желез
242