

4.Кетонурия и ее причины.
Кетоновые (ацетоновые) тела. В нормальной моче эти соединения встречаются лишь в самых ничтожных количествах (не более 0,01 г в сутки). При выделении больших количеств кетоновых тел качественные пробы становятся положительными. Это явление патологическое и называется кетонурией. Например, при сахарном диабете ежедневно может выделяться до 150 г кетоновых тел.
С мочой никогда не выделяется ацетон без ацетоуксусной кислоты, и наоборот. Обычные нитропруссидные пробы позволяют определить не только присутствие ацетона, но также и ацетоуксусной кислоты; β-оксимас-ляная кислота появляется в моче лишь при сильном увеличении количества кетоновых тел (сахарный диабет и др.).
Кетоновые тела выделяются с мочой не только при сахарном диабете, но и при голодании, исключении углеводов из пищи. Кетонурия наблюдается при заболеваниях, связанных с усиленным расходом углеводов: например, при тиреотоксикозе, кровоизлияниях в подпаутинные пространства, черепно-мозговых травмах. В раннем детском возрасте (продолжительные заболевания пищеварительного тракта (дизентерия, токсикозы) могут вызвать кетонемию и кетонурию в результате голода и истощения. Кетонурия нередко наблюдается при инфекционных заболеваниях: скарлатине, гриппе, туберкулезе, менингите. В этих случаях кетонурия не имеет диагностического значения и является вторичной.
Билет№28
Переваривание и всасывание нуклеопротеинов в ЖКТ. Судьба всосавшихся продуктов.
Переваривание нуклеопротеинов и всасывание продуктов их распада осуществляются в пищеварительном тракте. Под влиянием ферментов желудка, частично соляной кислоты, нуклеопротеины пищи распадаются на полипептиды и нуклеиновые кислоты; первые в кишечнике подвергаются гидролитическому расщеплению до свободных аминокислот. Распад нуклеиновых кислот происходит в тонкой кишке в основном гидролитическим путем под действием ДНК- и РНКазы панкреатического сока. Продуктами реакции при действии РНКазы являются пуриновые и пи-римидиновые мононуклеотиды, смесь ди- и тринуклеотидов и резистентные к действию РНКазы олигонуклеотиды. В результате действия ДНКазы образуются в основном динуклеотиды, олигонуклеотиды и небольшое количество мононуклеотидов. Полный гидролиз нуклеиновых кислот до стадии мононуклеотидов осуществляется, очевидно, другими, менее изученными ферментами (фосфодиэстеразами) слизистой оболочки кишечника.
В отношении дальнейшей судьбы мононуклеотидов существует два предположения. Считают, что мононуклеотиды в кишечнике под действием неспецифических фосфатаз (кислой и щелочной), которые гидролизируют фосфоэфирную связь мононуклеотида («нуклеотидазное» действие), расщепляются с образованием нуклеозидов и фосфорной кислоты и в таком виде всасываются. Согласно второму предположению, мононуклеотиды всасываются, а распад их происходит в клетках слизистой оболочки кишечника. Имеются также доказательства существования в стенке кишечника нуклеотидаз, катализирующих гидролитический распад моно-нуклеотидов. Дальнейший распад образовавшихся нуклеозидов осуществляется внутри клеток слизистой оболочки преимущественно фосфороли-тическим, а не гидролитическим путем.
Всасываются преимущественно нуклеозиды, и в таком виде часть азотистых оснований может быть использована для синтеза нуклеиновых кислот организма. Если происходит дальнейший распад нуклеозидов до свободных пуриновых и пиримидиновых оснований, то гуанин не используется для синтетических целей. Другие основания, как показывают опыты с меченными по азоту аденином и урацилом, в тканях могут включаться в состав нуклеиновых кислот. Однако экспериментальные данные свидетельствуют, что биосинтез азотистых оснований, входящих в состав нуклеиновых кислот органов и тканей, протекает преимущественно, если не целиком, de novo из низкомолекулярных азотистых и безазотистых предшественников.
Биосинтез триацилглицеринов, способы синтеза, последовательность реакций. Роль инсулина, адреналина, глюкагона в регуляции синтеза. Значение процесса.
Известно, что скорость биосинтеза жирных кислот во многом определяется скоростью образования триглицеридов и фосфолипидов, так как свободные жирные кислоты присутствуют в тканях и плазме крови в небольших количествах и в норме не накапливаются.
Синтез триглицеридов происходит из глицерина и жирных кислот (главным образом стеариновой, пальмитиновой и олеиновой). Путь биосинтеза триглицеридов в тканях протекает через образование α-глице-рофосфата (глицерол-3-фосфата) как промежуточного соединения.
В почках, а также в стенке кишечника, где активность фермента глицеролкиназы высока, глицерин фосфорилируется за счет АТФ с образованием глицерол-3-фосфата:
В жировой ткани и мышцах вследствие очень низкой активности глицеролкиназы образование глицерол-3-фосфата в основном связано с процессами гликолиза и гликогенолиза. Известно, что в процессе гли-колитического распада глюкозы образуется дигидроксиацетонфосфат (см. главу 10). Последний в присутствии цитоплазматической глицерол-3-фос-фатдегидрогеназы способен превращаться в глицерол-3-фосфат:
Отмечено, что если содержание глюкозы в жировой ткани понижено (например, при голодании), то образуется лишь незначительное количество глицерол-3-фосфата и освободившиеся в ходе липолиза свободные жирные кислоты не могут быть использованы для ресинтеза триглицеридов, поэтому жирные кислоты покидают жировую ткань. Напротив, активация гликолиза в жировой ткани способствует накоплению в ней триглицеридов, а также входящих в их состав жирных кислот. В печени наблюдаются оба пути образования глицерол-3-фосфата.
Образовавшийся тем или иным путем глицерол-3-фосфат последовательно ацилируется двумя молекулами КоА-производного жирной кислоты (т.е. «активными» формами жирной кислоты – ацил-КоА). В результате образуется фосфатидная кислота (фосфатидат):
Как отмечалось, ацилирование глицерол-3-фосфата протекает последовательно, т.е. в 2 этапа. Сначала глицерол-3-фосфат-ацилтрансфераза катализирует образование лизофосфатидата (1-ацилглицерол-3-фосфата, а затем 1-ацилглицерол-3-фосфат-ацилтрансфераза катализирует образование фосфатидата (1,2-диацилглицерол-3-фосфата) .
Далее фосфатидная кислота гидролизуется фосфатидат-фосфогидро-лазой до 1,2-диглицерида (1,2-диацилглицерола):
Затем 1,2-диглицерид ацилируется третьей молекулой ацил-КоА и превращается в триглицерид (триацилглицерол). Эта реакция катализируется диацилглицерол-ацилтрансферазой:
Синтез триглицеридов (триацилглицеролов) в тканях происходит с учетом двух путей образования глицерол-3-фосфата и возможности синтеза триглицеридов в стенке тонкой кишки из β-моноглицеридов, поступающих из полости кишечника в больших количествах после расщепления пищевых жиров.
Установлено, что большинство ферментов, участвующих в биосинтезе триглицеридов, находятся в эндоплазматическом ретикулуме, и только некоторые, например глицерол-3-фосфат-ацилтрансфераза,– в митохондриях.
Регуляция: Адреналин и норадреналин увеличивают скорость липолиза в жировой ткани; в результате усиливается мобилизация жирных кислот из жировых депо и повышается содержание неэстерифи-цированных жирных кислот в плазме крови. Как отмечалось, тканевые липазы (триглицеридлипаза) существуют в двух взаимопревращающихся формах, одна из которых фосфорилирована и каталитически активна, а другая – нефосфорилирована и неактивна. Адреналин стимулирует через аденилатциклазу синтез цАМФ. В свою очередь цАМФ активирует соответствующую протеинкиназу, которая способствует фосфорилированию липазы, т.е. образованию ее активной формы. Следует заметить, что действие глюкагона на липолитическую систему сходно с действием кате-холаминов.
Гормоны и их классификация. Представления об основных механизмах гормональной регуляции метаболизма.
Г – это химические посредники, регулирующие обмен в-в и развитие органзма.
Биологические признаки:
Дистантность действия
Строгая специфичность, т.е. один Г нельзя заменить другим
Высокая биологическая активность(в крови конц.10-6 – 10-11ммоль/л)
Г оказывают действие путями:
- изменяют проницаемость клеточных мембран
- изменяют скорость ферментативных р-ций – модулируют активность готовых мо-д Б-ферментов, это Г срочной регуляции(сек, мин)
- изменяют скорость синтеза Б-ферментов – Г медленной регуляции.
Классификация:
по химич.природе:
производные АК – адреналин(эпинефрин), норадреналин, тироксин и др.
пептидные(елковые) – АКТГ, ЛТГ, МЦС, инсулин
стероидные – половые, кортикостероиды.
по механизму передачи горм.сигнала в кл-мишени:
стероидные Г и тироксин. Их рецепторы располагаются в цитозоле клеток; они проникают во внутрь и реализуют гормональный эффект по цитозольному механизму.
пептидные и адреналин; рецепторы к этим Г на пов-ти клет.мембран, и Г не проникают вутрь клеток.
в звисимости от посредника:
Г реализуют свой эфф.через циклические нуклеотиды(цАМФ, цГМФ)
с участием ионизированного Са++ и инозитолполифостфатов (ИПФ)
Креатинурия и ее причины.
КРЕАТИНУРИЯ— появление креатина в моче. Моча здорового мужчины почти не содержит креатина, у женщин и детей — незначительная физиологическая креатинурия; у женщин она увеличивается при беременности, лактации. Избыточное содержание в пище мяса может привести к экзогенной креатинурией.
Основной причиной патологической креатинурии являются поражения мышц: миозиты, мышечная дистрофия, тяжелая миастения, тонические и клонические судороги.
Кроме того, креатинурия отмечается при ряде эндокринных заболеваний — диабете, гипертиреозе, акромегалии, а также при ацидозе, алкалозе, авитаминозах Е и С.
В норме образующийся в мышцах креатин ангидрируется н выделяется с мочой в виде креатинпна (около 2 г в сутки). Т. к. возможность организма ангидрировать креатин ограничена, возникновение креатинурии может иметь место как при повышенном распаде белка, так и при нарушении нормального превращения креатина в креатинин.
Определение креатина в моче производят путем превращения его в креатинин при помощи гидролиза 1 н. раствором соляной кислоты. Определяют количество креатинина в моче реакцией Яффе до и после гидролиза. Вычитанием первой величины из второй узнают содержание в моче креатина.
Билет№29
Пути распада пуриновых и пиримидиновых нуклеотидов в тканях. Конечные продукты. Нарушения обмена нуклеотидов. Биохимические основы подагры.
Превращение и всасывание липидов в желудочно-кишечном тракте.
Сут. потребность 180г. Должны быть представлены в разных кол-вах насыщенные, полиненасыщенные и мононасыщенные. 30% по калорийности от всей пищи. ХС в сут.должно быть менее 300мг.
рНопт. липазы жел.сока =5,5-7,5, а у взрос. 1,5, поэтому расщепление в желудке не происходит(в кишечнике!)
У груд.детей рНжел.сока 5,0. Л.расщепляются лингвальной липазой(рНопт 4,5)и желудочной липазой.
Основные фазы превращения:
Эмульгирование – образование из большиз липидных капель мелкодисперсной эмульсии(0,5мкм). Э.способствует: *натриевые сили желчных кислот, *СО2 – образ-ся при разложении бикарбонатов под действием соляной к., поступающей с пищевым комком из желудка, *образующиеся по мере расщеп.липидов мыла(соли жир.к-т и натрия R-COONa)
Осн.роль в эмульгировании у желчи – смеси натриевых солей желчных к-т, ФЛ и ХС желчных к-т в соотн. 12,5 : 2,5 : 1.
рН желчи больше 7,0.
Желчные к-ты – конечный продукт обмена ХС – производные холановой к-ты:
Желчные кислоты:
- первичные 3,7,12-триоксихолановая = холевая; 3,7-диоксиходановая = хенодезоксихолевая(образ.из печени из ХС)
- вторичные 3,12-диоксихолановая = дезоксихолевая; 3-оксихолановая=литохолевая(образ.в кишечнике под действием м/флоры)
**Конъюгирование желчных кислот: 1)с глицином – при у/в питании; 2)с таурином(образ.из цистеина)-при Б питании
роль: - эмульгируют жиры; -активируют липазу, -способсвуют всасыванию продуктов гидролиза жиров в кишечнике.
В сутки из ХС образ. 1г желчи в гепатоцитах→по печеночным пузырным протокам→концентр.в желч.пузыре.Опорожнение происходит постоянно, но усил.под действием поступившей пищи и ХЦкинина.
После выполенния своих ф.85%желчных кислот всас.в подвздошной кишке и вновь поступ.в состав секретируемой желчи – энтерогепатальный круг.
Общий пул желч.к-т 2,8-3,5г. Совершают 6-8 оборотов в сут.
Недостаток желч.к-т: желчекам.болезнь, стеаторрея.
Липолитическая фаза.
Осн.продукты – бета-моноацилглицерин и жир.к-ты.
Эфиры ХС(ферм.холестерол-эстераза панкреатич.и кишеч.соков) →жир+жир.к-ты.
Фосфатидилхолин (ф.панкреатич.фосфолипаза А2) →лизофосфолипид(панкреатич.лизофосфолипаза) →жир.к-та(из альфа)+глицерофосфохолин→всас.в кровь или выводятся.
Мицеллярная фаза. Транспортировка продуктов расщепления липидов от места образования к всас.пов-ти кишечника. Глицерин и жир.к-ты с Сменьше 10 всас-ся хорошо. Жир.к-ты с числом С-атомов больше 10, ХС, нерасщепленные ФЛ всас.с помощью мицелл. Они в сотни раз меньше эмульсий. В центрк гидрофобное ядро из жир.к-ты, моноглицерида и ХС. Снаружи гидрофильн.оболочка из ФЛ и жнлч.к-т.
мукозная фаза. Процессы ресинтеза в клетках эпителия кишечника – бета-моноглицеридный синтез ТАГ: поскольку жир.к-ты поступили извне(с пищей), из них образ.экзогенные:
R-COOH + HSKoA + АТФ (1)→ RCOSKoA + АМФ + ФФн
бета-МАГ + R1COSKoA(2) → ДАГ (3)→ ТАГ
Триглицеридсинтетаза: (1)ацилКоАсинтетаза, (2)моноглицеридацилтрансфераза, (3)диглицеридацилтрансфераза.
5) Транспортная фаза. Трансп.форма липидов – ЛП. Л, образованные в энтероцитах тонкогг киш. – экзогенные ТАГ, а также ФЛ и всосавшийся ХС – окружаются белком и формир. ХМ – очень крупные частицы, не способные проникать в кров. капилляры, поэтому диффундируют в димфатич.с-му кишечника→грудной лимфатич.проток. Легкие ограничивают поступление ХМ в арт.кровь(липопектическая ф.легких). ХМ не облад. атерогенностью: из-за больших размеров не проникают в капилляры. После приема пищи наблюдается гипергликемия(алиментарная) за счет ХМ. Х гидролизуются ЛП-липазой(крепится к стенкам сосудов гепарином)
Гормоны щитовидной и паращитовидной желез. Химическое строение и участие в обменных процессах.
тироксин. Фолликулярная часть щит.ж. 3,5,3’,5’-тетрайодтиронин
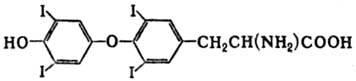
Регулирует скорость основного обмена, рост и дифференцировку тканей, обмен белков, у/ви липидов, водно-солевой обмен, деят-ть ЦНС, ЖКТ…
гипофункция: в детском возрасте – кретинизм, остановка роста, изменения кожи, волос. мышц, наруш.психики.
Гпотиреоидный отек(микседема). Слизистый отек, ожирение, выпад.волос и зубов, психич.расстройства.
Эндемический зоб(недостаток иода)
Гиперфункция: гипертиреоз – диффуз.токсич.зоб/Базедова болезнь: тахикардия, пучеглазие+зоб, общее истощение+психич.расстройства.
паратгормон. Паращитовид.ж. Белковая природа: 84 АК-остатка;состоит из 1 п/п цепи. Физиологическое влияние паратГ на клетки почек кост.ткани через систему аденилатциклаза-цАМФ. Регуляция конц.Са++ и связанных с ним анионов фосфотной к-ты в крови. В почках уменьш.реабс.Са++
Гипоф: тетанические судороги
Гиперф: вымывание солей Са в виде цитратов и фосфатов из костной ткани→деструкция мин.и орг. компонентов костей.
3) кальцитонин. С-клетки щит.ж. Пептидная природа. Выделяется в ответ на повышение Са++ в крови. Пост. конц. Са в крови → уменьш.резорбции кост.тк., гипокальциемия, гипофосфатемия – антагонист паратгормона.
Протеинурия и ее причины.
почечная
функциональная – временное проходящее нарушение почечного фильтра. Физиогогическая – у новорожд., обусловлена увелич. прониц-тью поч.фильтра; к 4-10 дню проходит. Ортостатическая(циклич.) – у детей 6-12 лет и подростков(приходящие изменения гемодинамики почки, например, из-за аномалии осанки). Инсулярная – у детей раннего возр.(повыш.раздражимость почесного фильтра)
органическая. Гломерулярная – поврежд.почеч.фильтра(селективная – в моче Б с массой менее 100кДа, и неселективная – в моче все Б крови). Тубулярная – поврежд.канальцев и наруш.работоспособности Б.
внепочечная
преренальная/перегрузочная – увелич. конц.общ.Б или появление парапротеинов в плазме: легкие цепи иммуноглоб(Б Бенс-Джонса), Нв(ППВ=1,25г/л), миоглобин(раюдомиолиз)
постренальная – попадание в мочу Б из мочевывод.путей при воспал.или опухолях.
Билет№30
Биосинтез ДНК. ДНК-полимеразы. Повреждения и репарация ДНК. Наследственные заболевания, связанные с нарушением репарации ДНК.