

Dale_Molecular Genetics of Bacteria 4th ed
.pdf
|
GENETIC MODIFICATION |
227 |
||
|
|
EcoRI |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cut with EcoRI
Ligate with insert DNA
EcoRI EcoRI
Insert
Figure 8.7 Structure and use of l insertion vectors
essential for lytic growth can be deleted from the vector to make room for the insert. However, if more than about 10 kb is removed, the size of the vector DNA falls below the minimum packaging limit, so that the vector itself would not be viable. This is unfortunate since one of the main uses of vectors, the construction of gene libraries (see below), often requires the cloning of fragments that are as large as possible.
Replacement vectors
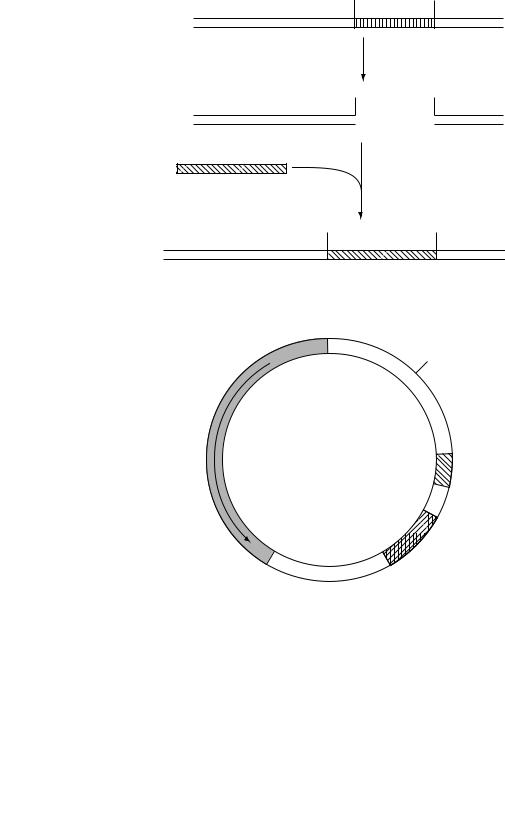
The cloning capacity of vectors can be increased by using a different approach. In the example shown in Figure 8.8, the vector contains two restriction sites rather than one. Cutting the DNA of this vector will therefore result in three fragments: the left and right arms and a central piece of DNA known as the stuffer fragment. The stuffer contains no genes needed for growth of the phage, so it can be separated and discarded. The left and right arms together are too small to allow viable phage particles to form; such particles require an additional inserted piece of DNA which must be, in this case, at least 7 kb and not more than 22 kb. Therefore, not only is the cloning capacity increased, but such vectors are selective for reasonably large inserts. Vectors of this type are known as replacement vectors.
8.4.5 Cloning larger fragments
Packaging of DNA requires only the DNA region around the cos sites (see Chapter 4). It is therefore possible to use in vitro packaging with a class of vectors known as cosmids (Figure 8.9). These are simply plasmids into which a cos site has
228 |
MOLECULAR GENETICS OF BACTERIA |
||
|
|
BamHI |
BamHI |
|
Left arm (22 kb) |
Stuffer (11 kb) Right arm (8 kb) |
|
|
|
|
Cut with BamHI |
|
|
|
Remove stuffer fragment |
|
|
BamHI |
BamHI |
|
Left arm |
|
Right arm |
|
Insert |
|
|
|
|
Add insert fragment |
|
|
|
and ligate |
|
|
BamHI |
BamHI |
|
|
Left arm |
|
Right arm |
|
|
Insert size is between |
|
|
|
7 and 22 kb |
|
|
Figure 8.8 Structure and use of l replacement vectors |
||
BamHI |
bla |
cos |
site |
ori |
Figure 8.9 Structure of a cosmid. ori, origin of replication; bla, b-lactamase (ampicillin resistance as a selectable marker); BamH1, restriction enzyme recognition site where DNA can be inserted. The presence of the cos site enables the plasmid to be packaged into l phage particles provided that a sufficiently large piece of DNA has been inserted
been introduced. The cosmid itself is much too small to be packaged successfully. However, if the insert is large (say 40 kb), then the recombinant cosmid will be large enough for viable particles to be produced. These particles can then infect a
GENETIC MODIFICATION |
229 |
sensitive host, but in this case antibiotic-resistant colonies rather than phage plaques will indicate the success of the procedure.
Larger fragments can be cloned using vectors based on the bacteriophage P1, which have a cloning capacity of 95 kb, or the so-called bacterial artificial chromosome (BAC) system which is based on the F plasmid and is able to maintain inserts of greater than 300 kb. These vectors have been particularly useful in genome sequencing projects (Chapter 10).
8.4.6 Bacteriophage M13 vectors
The filamentous bacteriophage M13 (see Chapter 4) provides the basis for another group of vectors. With M13, the phage particles contain single-stranded DNA while the replicative form is double stranded. Fragments of DNA can therefore be inserted into the replicative form of the M13 DNA. Transformation of a suitable host will produce plaques from which the recombinant phage can be obtained. Since the phage particles contain single-stranded DNA, this is a convenient way of obtaining cloned DNA in a single-stranded form, which is useful for some applications such as site-directed mutagenesis. Gene sequencing originally depended heavily on obtaining single-stranded templates using M13 vectors. Although modern sequencing methods can use double-stranded templates, M13 clones are still occasionally used for this purpose.
8.5 Gene libraries
8.5.1 Construction of genomic libraries
A gene library (or more precisely a genomic library, to contrast it with a cDNA library – see below) is a collection of recombinant clones each of which carries a different piece of DNA from the organism of interest, so that between them they represent the complete genome of that organism.
To construct a genomic library, the DNA from the source organism is fragmented randomly and the mixture of pieces joined to the vector DNA (see Figure 8.10). This will produce a number of recombinant molecules, each having a different piece of source DNA. No attempt is made to separate these molecules; instead the whole mixture is used for transformation of E. coli. Each individual clone carries a different piece of DNA and the whole collection of clones constitutes the gene library.
The number of clones needed for a complete genomic library depends on the size of the genome and the average size of fragment cloned. For example, for a bacterium with a genome of 4 106 base pairs and an average insert size of 18 kb (using a vector), a library of 1000 clones gives a 99 per cent probability of

230 MOLECULAR GENETICS OF BACTERIA
EcoRI
Vector,
cut with EcoRI
Random mixture of
DNA fragments
Mix and ligate
Mixture of recombinant DNA molecules
Transformation
Collection of bacterial clones constitutes a gene library
Figure 8.10 Construction of a genomic library using a plasmid vector
recovering any specific gene. With an average insert size of say 1 kb, about 18 000 clones would be required to achieve 99 per cent representation of the same genome. (The calculation has to take account of the overlap and duplication that will occur in a random library). Using the techniques described in the next section, it is not difficult to screen several thousand clones, so even a small insert library is quite usable for a bacterial genome. But with larger genomes, the need
GENETIC MODIFICATION |
231 |
for larger inserts becomes important, unless machines are available which can handle hundreds of thousands of clones.
8.5.2 Screening a gene library
Making a genomic library of bacterial DNA is comparatively straightforward. However, it is necessary to have some way of finding what is required. Rapid screening methods have been developed to enable large numbers of colonies to be tested simultaneously, commonly using either gene probes or antibodies.
Screening with gene probes
The use of gene probes relies on the hybridization of complementary singlestranded nucleic acid sequences (see Chapter 1). Screening a gene library involves transferring a print of the colonies (or phage plaques) to a filter, which is subsequently hybridized with a labelled gene probe. A variety of non-isotopic labels are now commonly used, rather than radioactive labels. The position of a positive reaction on the filter can be correlated with the position of a specific clone which can be picked off and purified.
The main factor is how to obtain the required probe. If it is known, or suspected, that the gene required is related to one that has already been characterized from another source, then that DNA sequence can be used as a heterologous probe, using a low stringency of hybridization (to allow for a degree of mismatching between the DNA sequences).
Alternatively, if a small amount of purified protein can be isolated, then it is possible to determine the amino acid sequence of a small part of that protein (usually the N-terminal sequence). From that information, the likely sequence of the DNA can be deduced and a short probe corresponding to that sequence can be synthesized.
Polymerase chain reaction
An alternative way to generate the required probe is by using the polymerase chain reaction (PCR; see Chapter 2). For this purpose, the PCR primers are derived from the sequence of related genes in the databanks, using portions of the genes that are very similar in all known sequences of this family of genes (conserved regions). If the target gene has sufficiently similar sequences, the primers will anneal and the PCR will therefore amplify a DNA fragment from that gene (Figure 8.11). This product can then be labelled and used to screen a genomic library.

232 |
MOLECULAR GENETICS OF BACTERIA |
|
Conserved regions |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
B |
genes |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
Known |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Consensus
PCR primers
Unknown
PCR
Probe
Screen gene library
Figure 8.11 Use of PCR to generate a probe for screening a gene library. Related genes A–D, from different sources, have two conserved regions (i.e. they are similar in all four genes). PCR primers designed to recognize these conserved sequences can be used to amplify part of the corresponding gene from another source. The amplified product can be used as a probe for screening a gene library so as to isolate the complete gene
The use of PCR can bypass the need for making a gene library at all. If we are studying a specific gene in closely related species and the sequence of the gene from one species is known, then the primer sequences from the known gene can be simply derived and used to amplify the corresponding gene from the other organisms. This application will be considered further in Chapter 10.
Screening gene libraries with antibodies
If antibodies to the protein of interest can be produced, these can be used to screen a gene library in much the same way as described for a gene probe. There are certain limitations to this approach: the gene concerned must be expressed (although not
GENETIC MODIFICATION |
233 |
necessarily in an active form), the antibody should recognize the protein in a denatured state (and hence should recognize a linear epitope rather than a conformational one) and it must recognize the primary product of translation rather than some modified form (e.g. a glycosylated epitope), since this post-translational modification may not occur in the recombinant bacterium. Despite these limitations, antibody screening has been widely used, especially for genes coding for key antigens of specific pathogens (see the description of gt11 below).
Screening gene libraries by complementation
It is also possible to screen a gene library by testing for the ability to complement specific mutant strains of E. coli. For example, a leucine auxotroph (which is unable to grow without added leucine) may be converted to prototrophy by a recombinant plasmid carrying a leu gene from another bacterium. The required clone can therefore be identified by plating the library onto a medium lacking leucine.
Of course, this procedure only works if the cloned gene is expressed and only if the product is functional (and readily detected) in E. coli. It is therefore not the most powerful way of screening gene libraries, but it is particularly useful for confirming the identity of cloned genes when a possible function has been identified by sequence comparisons.
8.5.3 Construction of a cDNA library
In some circumstances, it is advantageous to make a gene library from the mRNA rather than from the DNA. This requires the synthesis of double-stranded DNA using the mRNA as a template. There are many variations in the details of this procedure, which will not be considered here, but the central feature is the use of an enzyme known as reverse transcriptase (since it is an RNA-directed DNA polymerase, as opposed to the enzyme responsible for transcription proper which is a DNA-directed RNA polymerase). The DNA produced (complementary DNA, or cDNA) can be ligated to a vector to produce a cDNA library.
Libraries of cDNA are commonly used for animals and other eukaryotes. Since a cDNA library contains only those genes that are expressed in the starting material, it will be much smaller than a genomic library. Furthermore, eukaryotic genes usually contain introns (see Chapter 1); these are removed during processing of the mRNA, so the size of a cDNA clone needed to contain the complete gene is much smaller than the corresponding length of genomic DNA.
Bacterial cDNA libraries are less commonly used. Since bacterial genomes are smaller, and bacterial genes do not often contain introns, there is less of an advantage in using a cDNA library (and making a genomic library is much
234 |
MOLECULAR GENETICS OF BACTERIA |
easier). Nevertheless, bacterial cDNA libraries are used occasionally, for example to identify and isolate genes that are selectively expressed under specific growth conditions. Other applications of reverse transcriptase will be discussed in Chapter 10.
8.6 Products from cloned genes
8.6.1 Expression vectors
If a gene from one organism is put into another, it may not be expressed very well if at all. The most common reason for this is that the promoter may not be recognized by the RNA polymerase of the new host. So, if expression of the product is required, an expression vector (see Figure 8.12) would be used which contains an E. coli promoter adjacent to the cloning site. Insertion of a DNA fragment at this site, in the correct orientation, places it under the control of the promoter provided by the vector, thereby ensuring expression in the recombinant
E. coli clone.
A further development of this concept is to use the vector to supply translational signals as well as a promoter. The result (if the construct is designed to maintain the correct reading frame) will be a fusion protein which has an N-terminal sequence provided by the vector and a C-terminal region which is the product that is to be detected. This can improve the expression and stability of the product.
Often once a protein has been expressed in E. coli, the next step would be to purify the protein and separate it from the other proteins present in the cell. Protein purification can be simplified by using specialized expression vectors that incorporate an affinity tag at the N-terminal sequence of the cloned protein. Peptides with short stretches of histidine residues bind specifically to nickel, for example. If the gene is manipulated so that the protein has a string of histidines (usually six to 10) at its N-terminus, the His-tagged protein can be purified on a column containing a solid phase with bound nickel ions.
l gt11
One example of an expression vector, although used in a rather different way, is the vector gt11 (Figure 8.13). This is an insertion vector with a single EcoRI site within a b-galactosidase gene, so that an inserted DNA fragment will be under the control of the lac promoter. Furthermore, if the insert is in the correct reading frame, the peptide coded for by that fragment is actually linked to the b-galacto- sidase. This vector is useful for gene libraries that are to be screened with antibodies, for example in the identification and isolation of genes coding for protein antigens of pathogenic bacteria.

GENETIC MODIFICATION |
235 |
Cloning |
site |
Promoter |
|
bla |
|
ori |
|
Insert required gene
|
Gene |
|
product |
Inserted |
|
gene |
|
bla |
|
ori |
ori |
Figure 8.12 Basic features and use of an expression vector. The cloning site is adjacent to a promoter on the vector so that inserted DNA will be transcribed from that promoter. With some expression vectors, translational signals are also included
Maximizing product formation
The application of gene cloning which received most publicity, before the advent of gene therapy and transgenic animals, was the production of foreign proteins by microbial hosts. For example, human growth hormone (HGH or somatotropin) is a polypeptide hormone which is used to treat the condition known as pituitary dwarfism. Prior to gene cloning, the only source of this material was human pituitary glands removed at autopsy. The limitations on supply coupled with

236 |
|
|
|
MOLECULAR GENETICS OF BACTERIA |
||||
|
|
(a) Vector |
|
|||||
|
|
|
|
|
P |
|
Eco RI |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
β-galactosidase |
|
|
|
|
|
|
|
|
||
|
|
(b) Recombinant |
Eco RI |
|||||
|
|
|
|
|
|
|
Eco RI |
|
P
Insert
Figure 8.13 Use of l gt11 for generation of fusion proteins. (a) The vector carries a b-galactosidase gene with an EcoRI site within it. (b) Insertion of DNA at the EcoRI site disrupts the lacZ gene. Functional b-galactosidase is not made but (if the insert is in the correct reading frame) a fusion protein is produced
the risks of transmission of latent infections, were a powerful stimulus for efforts to develop an alternative supply. Cloning and expressing the gene in E. coli provided a safe and plentiful supply of the hormone.
In order to take full advantage of bacterial expression of foreign proteins, sophisticated expression vectors are used to maximize yields of the recombinant product. Extremely high levels of specific protein, representing perhaps 50 per cent of total cell protein, can be produced using such a vector. However, diverting so much of the cell’s resources into a product that is useless as far as the cell is concerned will slow down the growth rate (which will also exacerbate plasmid instability as discussed in Chapter 5). The usual strategy for overcoming this problem is to use a promoter that is subject to repression so that the bacteria can be grown to high cell density before product formation is induced by altering the growth conditions. For further details and specific examples, see the reading list in Appendix A.
8.6. 2 Making new genes
DNA synthesis
Gene cloning need not be restricted to DNA sequences which occur in nature. It is possible to synthesize stretches of DNA (oligonucleotides) of any sequence using a DNA synthesizer. The operator merely types in the required sequence and the machine does the rest. Since this is a sequential process, the accuracy tends to decline with longer oligomers, but sequences of up to 100 bases can be constructed with a reasonable degree of accuracy. Longer sequences can be made by enzymatic joining of these shorter oligomers.