

Thermal Analysis of Polymeric Materials
.pdf3.4 Copolymerization and Reactions of Polymers |
227 |
___________________________________________________________________
properties of all known polymers and the development of prediction schemes. Most well-known are the property tables of van Krevelen, based on the addition of contributions from the constituent chemical groups [21]. If the connection between properties and chemical structure is not fully known, it is still possible to predict the properties using neural network computations as shown in Appendix 4 [22,23]. With such networks it is possible to develop predictions based on all present knowledge in an area. A prediction of properties of copolymers with neural network techniques has been described [24].
3.4.1 Chain Reaction Copolymers
The kinetic propagation reaction equations for radical copolymerization of two monomers, M1 and M2, are written in Fig. 3.45. The complications due to additional, different monomers, as well as transfer, reverse, and termination reactions increase the numbers of equations and rate constants, so that the resulting reaction equations are unhandy, and it needs much experimental work to establish the rate constants. Also, the computational effort to solve the many equations becomes excessive.
Fig. 3.45
For the two different monomers, there are four propagation reactions in Fig. 3.45. The chance that M1•, the free radical terminating a chain ending in M1, reacts with the same monomer M1 is P11. The chance to continue with monomer M2, is P12 which equals (1 P11). The analogous probabilities for M2• are P22 and P12 (= 1 P22). The probabilities are equal to the rates of the appropriate reaction divided by the sum of both possible rates, as indicated in Fig. 3.45 for P11. Three more definitions are given in Fig. 3.45, two for the reactivity ratios, r1, and, r2, and the third, for the monomer ratio x, also called the feed ratio (x = f1/f2). The basic copolymer equation is shown in the box at the bottom, where d[M1]/d[M2] = F1/F2 is the composition ratio.

228 3 Dynamics of Chemical and Phase Changes
___________________________________________________________________
The copolymer equation is derived for the steady state when both, [M1•] and [M2•] do not change with time. Steady state requires also that there are the same number of conversions of molecular radical ends M1• to M2• and vice versa, and there are equal rates of change of M1• to M2• and vice versa, i.e., the second and third rate expressions of Fig. 3.45 must be equal: k12[M1•][M2] = k21[M2•][M1].
Dividing the top rates of Fig. 3.45, considering r1 = k11/k12 and r2 = k22/k21 and using the equality, above, the ratio d[M1]/d[M2] is the copolymer equation, looked for:
The copolymer equation represents the composition ratio of the copolymer produced at the given instant and is rewritten in Fig. 3.46. The final correlation between composition ratio, F1/F2, and the feed ratio, x = f1/f2 = [M1]/[M2], can be reorganized, as also given in Fig. 3.46. For a reaction in a continuous flow reactor in steady state, one can thus predict the composition of the copolymer from the monomer feed ratio. In a batch reaction, in contrast, the composition changes continuously.
Fig. 3.46
In the plots included in Fig. 3.46 some typical copolymer-feed/composition diagrams are compared. The horizontal line of example 1 represents the case of copolymerization when both reactivity ratios are zero. Irrespective of the feed composition, the polymer composition has a 0.5 mole fraction of both components. The reaction leads to a strictly alternating copolymer since M1• cannot be followed by M1 (r1 = 0). The same holds for M2• (r2 = 0). A copolymer that fulfills this
3.4 Copolymerization and Reactions of Polymers |
229 |
___________________________________________________________________ |
|
requirement can be made of tetrafluoroethylene, |
CF2=CF2, and isobutylene, |
CH2=C(CH3)2.
The kinetics of example 2 shows no preference between the two monomers. The resulting copolymer is azeotropic, meaning that feed and composition are always equal (F1 = f1,). An example is found in 1,3-butadiene, CH2=CH CH=CH2, polymerizing with isoprene, CH2=CCH3 CH=CH2.
Random copolymers are always found if the product r1×r2 = 1, i.e., both monomers have the same preference for monomer 1 over 2 (k11/k12 = k21/k22). The examples 3a and 3b are two possible random copolymers with opposite reactivity-ratios r1 and r2. A typical random copolymer can be made of 1,3-butadiene, CH2=CH CH=CH2, and styrene, CH2=CH C6H5).
To produce a copolymer with alternating tendency, the product r1×r2 must be < 1. The examples 4 are two typical representatives. Note the tendency to approach curve 1 when f1 is 0.4 in case 4a, and f2 = 0, in case 4b. An example copolymerization of this type is methyl methacrylate, CH2=CCH3 CO O CH3, copolymerized with styrene, CH2=CH C6H5.
Example 5 in Fig. 3.46, finally, is characteristic for a tendency to make blocky copolymers. In this case r1×r2 >> 1, as is found in poly(propylene-co-styrene). Once a given radical is formed, it prefers to add the same monomer rather than to switch to the other.
In all these cases the copolymers are not precisely of the same composition and sequence lengths from molecule to molecule, as could result from a stepwise reaction (see Sect. 3.1). Distributions of sequence lengths exist in all these copolymers and could, again, be calculated from the reactivity ratios. Figure 3.46 summarizes the wide variety of composition diagrams possible.
3.4.2 Step Reaction Copolymers
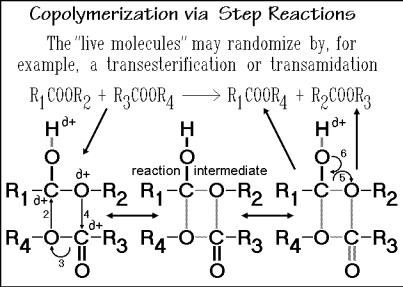
Step reaction copolymers tend to be more or less random. Even with different monomer reactivities that lead initially to non-random copolymers, randomization is ultimately helped by interchange reactions that can proceed after the initial polymerization. Figure 3.47 illustrates the interchange of chain segments by transesterification. One molecule has the chain-ends R1 and R2, the other R3 and R4. After the reaction, the ends are exchanged.
Three stages of the reaction-sequence are shown at the bottom of the figure which are similar to the initial polymerization shown in Fig. 3.14. The slow parts of the reaction involve the molecular movements. First is the addition of the catalyst, such as H+ which initiates the shift in the electron configuration, and the approach of the two chains to be interchanged. The electronic rearrangements, labeled 2, 3 and 4, are fast, lead to a distribution of the charge, +, and ultimately produce the reaction intermediate with a rather symmetric partial bonding. On removal of the original H+, the molecules may separate either with their old structure, or, with electron shifts 5 and 6 to the new molecules R1 COO R4 and R2 COO R3, as shown. In this way, it is also possible to produce random copolymers by melting the homopolymer components and keeping them for a sufficiently long time at high temperature to interchange the repeating units. This type of reaction is of importance to remember when
230 3 Dynamics of Chemical and Phase Changes
___________________________________________________________________
Fig. 3.47
discussing compatibility of step-reaction polymers in Chap. 7. If interchange reactions are possible, compatibility may be achieved by forming a copolymer. Similarly, an initially random copolymer may develop blocky sequences if one of the components may preferentially crystallize at a temperature where ester-interchange is possible. Crystallization removes the longer sequences of ' identical repeating units with a mole fraction if n' = P11' 1(1 P11). The interchange reaction can, then, recover these sequences, which, in turn, permits further crystallization and leads to a blocky copolymer. Homopolymers can, similarly, remove hindrances to crystallization by interchange reactions on folds and tie molecules of crystal surfaces discussed in Chap. 5. This interchange reaction limits the integrity of the molecules that was assumed to exist in flexible, linear macromolecules and points thus at the existence of intermediate compounds in the classification scheme of molecules of Sect. 1.1.3.
3.4.3 Gelation
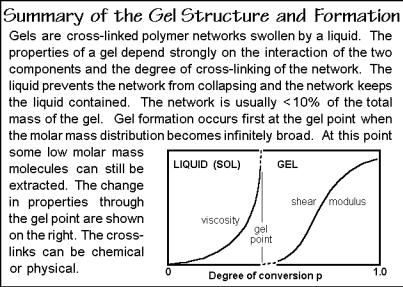
Polymerizations lead to linear macromolecules only if the monomers are strictly bifunctional. Monofunctional molecules were shown in Fig. 3.13 to limit the molecular length of step-growth polymers. Adding monomers with a higher functionality than, f = 2 to bifunctional monomers leads to network formation with structures as discussed in Sect. 1.2. A sudden gel formation is illustrated in Fig 3.48. Gel formation is marked by the appearance of a macroscopic network. The network has a practically infinite molar mass. In polymerizations from the pure monomer the network is swollen by monomer, oligomers, and linear and branched polymer molecule. In polymerization from solution, the solvent makes up a large number of the small molecules. As the polymerization continues, most or all of the polymerizable material joins the network. A gel can hold a large number of small
3.4 Copolymerization and Reactions of Polymers |
231 |
___________________________________________________________________
Fig. 3.48
molecules, such as monomers and/or solvents, because of the large volume of a coiled macromolecule and its low-mass density which was estimated from the random coil dimensions calculated in Sect. 1.3.
The gel formation can be linked to the functionality, f, and a branching coefficient,. The branching coefficient gives the probability that a specific functional group (of functionality > 2) is connected to another branch point. One can deduce from statistical reasoning that a gel appears at a critical branching coefficient, c. For f = 3, a gel appears when c is 0.5, i.e., there is a 50% chance that each branch is connected to another branch point. The network structure depends on the concentration of branch points and the degree of polymerization.
Gels can also form by physical cross-linking. Most common are the entanglements in a concentrated polymer solution that can lead to a gel. Further enhancement of gel formation is possible by bonding between the molecules by hydrogen bonding or polar interaction.
Special gelation is based on phase separation within polymer solutions. If the separating phase exists as small, solid particles (crystal or glass), it may represent a cross-link of very high functionality. Even the typical semicrystalline polymer can be characterized as tightly cross-linked, nanophase-separated gels, as will be discussed in Chap. 5.
3.4.4 Decomposition
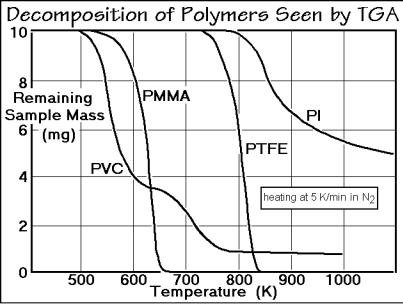
Decomposition can be looked upon as the reverse reaction of synthesis. Polymers with a ceiling temperature can simply show a reverse of the polymerization reaction as seen in Fig. 3.30. Figure 3.49 represents typical thermogravimetry traces for the decomposition of poly(methyl methacrylate), PMMA, and polytetrafluoroethylene,
232 3 Dynamics of Chemical and Phase Changes
___________________________________________________________________
Fig. 3.49
PTFE. Both polymers yield mainly monomer on heating. Note that this process is nothing else but the sublimation or evaporation, common when heating small molecules or rigid macromolecules. In fact, PTFE is a good laboratory source of tetrafluoroethylene which is pure enough to be repolymerized on a cold surface, particularly in the presence of a free radical catalyst.
The reversal of polymerization, however, is not the norm for polymer decomposition. Most polymers show decomposition to multiple products. Especially in the presence of oxygen is there always a chance of oxidation, or even self-sustaining burning. Such reactions are better called pyrolysis than decomposition. The thermogravimetry is for this reason usually done in an inert atmosphere, such as nitrogen, unless oxidation is to be studied.
The poly(vinyl chloride), PVC, of Fig. 3.49 decomposes in two steps. In the first it loses most of its chloride as HCl between two neighboring carbon atoms:
CH2 CHCl CH2 CHCl CH2 CHCl CH2 CH=CH CHCl CH=CH + 2HCl
In the second pyrolysis step, the polymer breaks up into many small molecules. (Note that the Cl trapped by two double bonds, as shown in the reaction sequence, above, cannot react as easily as a Cl next to a CH2-group as exists in uninterrupted sequences of CH2 CHCl ). The remaining mass after pyrolysis consists of highly cross-linked carbon in the form of char.
The aromatic polyimide (PI) of Fig. 3.49 is a relatively stable polymer, such as a polypyromellitimide, that is usable for high-temperature applications such as needed for insulation. It finally decomposes to small fragments that carry away most H-atoms and leave highly cross-linked, high-carbon chars.
3.4 Copolymerization and Reactions of Polymers |
233 |
___________________________________________________________________
3.4.5 Polymer Reactions
Nine examples of reactions after polymerization are selected in this section. Further details of these industrially important reactions should be looked-up in the General References given in the Preface, as is also proposed for all other examples of materials mentioned in this book, and partially illustrated for the following reactions.
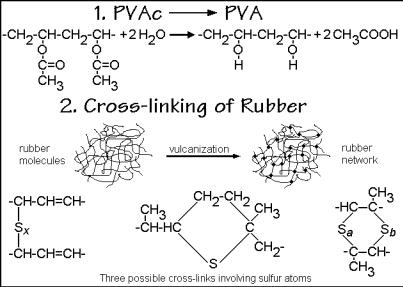
The first example is the well-known conversion of the easily polymerized poly(vinyl acetate), PVAc, CH2 CH(OCOCH3) , by hydrolysis to the technologically important poly(vinyl alcohol), PVA, CH2 CHOH . Figure 3.50 lists the chemical
Fig. 3.50
equation of the process. Poly(vinyl acetate), the reactant, is an amorphous polymer with a glass transition temperature of 305 K. It is produced in megaton quantities and used as polymerized for emulsions, water-based paints, adhesives, and paper coatings. Its monomer is made by oxidative addition of acetic acid to ethylene and polymerized using free radical initiators in bulk, solution, or emulsion. Dependent on molar mass, the polymer may be a viscous liquid or brittle solid. It is not soluble in water, but in ethanol, propanol, or butanol containing 5–10% of water. It is water-sensitive, but does not hydrolyze at a neutral pH. The product of the hydrolysis, the poly(vinyl alcohol), is the largest-volume, synthetic, water-soluble polymer. Its main uses are in fibers, adhesives, production of poly(vinyl butyral) described in Fig. 1.14, textile and paper sizing, cold-water soluble bags, films, and packaging, water thickening agents and drag reducers. The properties of PVA vary considerably with the production conditions. The crystals have a melting temperature of 538 K and the amorphous PVA has a glass transition temperature of 358 K. The polymer is solvent, oil, and grease resistant and has a high tensile strength and good oxygen barrier properties when dry.

234 3 Dynamics of Chemical and Phase Changes
___________________________________________________________________
The second example of a polymer reaction is the industrial cross-linking of rubber by vulcanization sketched in Fig. 3.50. The process was invented already in 1839 by C. N. Goodyear1 without knowledge of its chemical structure. Natural rubber is cis- poly(1-methyl-1-butenylene) or polyisoprene with a low glass transition temperature of about 210 K. Its structure and those of other rubbers are given in Fig. 1.15. The addition of sulfur in the form of S8 rings and heating causes the vulcanization. Of the listed cross-links in Fig. 3.50, only the left example is an efficient network former. The sulfur introduces about 1 cross-link for each of 50 S-atoms used. Modern vulcanization involves activators and accelerators for increased efficiency. The detailed mechanism is rather complicated and not fully understood.
Other cross-linking reactions are shown in Fig. 3.51. They can lead to products that can be first shaped by melting the flexible macromolecules (plastics) and then fixing the shape by cross-linking, approaching a rigid macromolecule as seen in
Fig. 3.51
Fig. 1.6. Reaction 3 involves ionizing radiation in form of X-rays, gamma rays, or electrons which impart high energy to a polymer to break bonds, producing ionic or free radical fragments of the molecules. The most common radiation sources are 60Co for gamma rays of 1.25 MeV and electron accelerators (0.1–10 MeV). These energies compare to typical bond energies of only 100 to 1000 kJ (= 1–10 eV). Reaction 3 illustrates the cross-linking of polyethylene, started by the production of free radicals (top reaction). Two free radicals of different molecules can then combine in a crosslink, as seen in the second reaction. The cross-linking is most efficient in the liquid state in order to have enough mobility in the chain segments to be combined.
1 Charles N. Goodyear, 1800–1860. The American inventor of the vulcanization cross-linking process which made possible the commercial use of rubber as one knows it today.

3.4 Copolymerization and Reactions of Polymers |
235 |
___________________________________________________________________
Semicrystalline polyethylenes cross-link only in the amorphous regions. After sufficient cross-linking, the polymer does not flow any more. Typical polymers that can be cross-linked similarly are polypropylene, polystyrene, polyacrylates, and poly(vinyl chloride). Uses of the cross-linked polymer include wire and cable insulation, heat-shrinkable films and tubing.
Polyacrylonitrile, PAN, CH2 CH(C N) , leads on pyrolysis over an intermediate stage to complete carbonization, as depicted in Fig. 3.51. The final product is used to make strong carbon fibers. The polyacrylonitrile fibers turn yellow to red on the formation of the ladder structure shown in the figure. On final carbonization the fiber turns black and consists of graphite-like structures. The carbon fibers have found many applications due to their high tensile modulus in the fiber direction and their, compared to metals, low weight. They are used as fiber reinforcement in epoxy matrices. Typical applications range from aircraft and aerospace to industrial, transportation, and recreational equipment.
The fifth polymer reaction is an example of grafting to structures described in Sect. 1.2. This reaction can be used to change the nature of a macromolecule by adding side-chains as shown schematically in Fig. 3.52. It allows, for example, compatibilization with other polymers (see Chap. 7) or the attachment of groups that
Fig. 3.52
change the physical properties. The grafted side-chain may be thermodynamically incompatible with the main-chain and leads then to multi-phase polymers. The morphology of the heterophase polymers is composition dependent and may also be affected by thermal history and precipitation conditions. Typical examples of graft copolymers are high-impact polystyrene (HIPS, styrene, polymerized in the presence of rubber, such as 1,4-polybutadiene), acrylonitrile-butadiene-styrene copolymers (ABS), and cellulose grafts. The ABS example is done by free radical polymerization
236 3 Dynamics of Chemical and Phase Changes
___________________________________________________________________
of acrylonitrile and styrene in the presence of 1,4-polybutadiene, as shown in Fig. 3.52. Free radicals attack on the remaining double bonds 1,4-polybutadiene crates graft sites for the copolymerization. Some free copolymer forms besides the graft.
The bottom reaction of Fig. 3.52 involves the chemical etching of semicrystalline hydrocarbons such as polyethylene and polypropylene. The surfaces, including the amorphous sections of the molecules are degraded oxidatively to low-mass fragments using, for example, ozone, permanganates, or nitric acid. The fold surfaces and strained sections in the amorphous fraction are broken and transformed into oxidized chain ends. The amorphous parts and surfaces of a semicrystalline polymer are, thus, removed first and a microcrystalline powder remains and can be analyzed more easily than when surrounded by amorphous material. The microcrystalline powders have found industrial applications. This reaction is also used to oxidize nonpolar polymer films to make them hydrophilic, so that they can be wetted for printing. Similar to the hydrocarbons, the molecular backbone of polyesters and polyamides can be broken by hydrolysis. These reactions can be done in the presence of water at elevated temperature with or without a catalyst. Contrasting these degradations are dissolutions with solvents, which lead to removal of whole molecules from the sample.
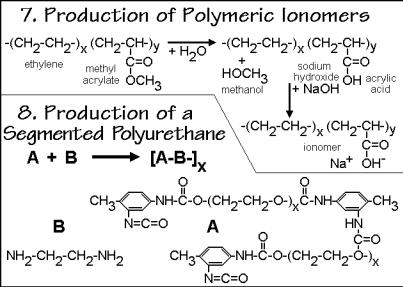
Ionomer production involving saponification of ester side-chains, such as in polyacrylates in the presence of NaOH, are shown in Fig. 3.53 as polymer reaction 7. The
Fig. 3.53
pendent ionic groups interact to form ion-rich nanophases dispersed in the nonpolar polymer matrix and create a cross-linked system. Typical ionomers are copolymers with ionized groups on about 1–10% of the repeating units. A common route to the ionomer is the modification of the preformed polymer, such as the introduction of a sulfonate group in polystyrene, or as shown in the example for reaction 7, the saponification of the poly(methyl acrylate-co-ethylene) followed by neutralization of