Дизметаболические нефропатии у детей
Кафедра госпитальной педиатрии
В структуре нефропатий у детей преобладают заболевания врождённого и наследственного генеза, а также болезни, связанные с наследственной предрасположенностью, имеющие скрытое начало и торпидное течение, среди которых наиболее значительную группу составляют пациенты с обменными нефропатиями.

Кристаллурия
•Кристаллурия – мочевой синдром, при котором в моче выявляются кристаллы различных солей (ОАМ с микроскопией осадка!!!)
•Часто носит транзиторный характер (лихорадка, респираторно-вирусные инфекции).
•При выздоровлении уменьшается или исчезает.

• Дизметаболические (обменные) нефропатии - группа заболеваний почек с различной этиологией и патогенезом, которые характеризуются поражением интерстициальной ткани почек и патологии канальцев, вследствие нарушения обмена веществ.

•Продукты нарушения обмена
приводят к:
•изменениям функционального состояния почек
•структурным сдвигам на уровне различных элементов нефрона, за счёт их накопления в почечной ткани, так и вследствие прямого токсического воздействия на канальца и интерстициальную ткань

Структура кристаллурий и ДНП
•Кальций 70-90%
•85-90% случаев - оксалат кальция
•3-10% случаев - фосфат кальция или смешанные
•5% случаев - ураты
•3% случаев - цистин
•5-15% случаев трипельфосфаты (фосфаты, аммоний, магний и кальций)

Факторы риска
•Отягощённый генеалогический анамнез (МКБ, ЖКБ, ожирение, гиперлипидемия, сахарный диабет)
•Гипоксия во внутриутробном периоде
•Заболевания желудочно-кишечного тракта, эндокринопатии
•Неправильное питание
•Недостаточное употребление жидкости

• Первичные дизметаболические нефропатии являются наследственно -
обусловленными заболеваниями, характеризуются торпидным течением, ранним развитием уролитиаза, хроническим ТИН и прогрессированием в хроническую почечную
недостаточность.
• Первичная наследственная гипероксалурия
(оксалоз 1 и2 типа), Синдром Леша-Нихана, нефропатический цистиноз.

Оксалоз 1и 2типа
•Аутосомно рецессивный или доминантный тип наследования
•Моногенная мутация – нет фермента для обмена глиоксиловой кислоты - усиление внепочечного синтеза оксалатов
•Кристаллы откладываются во во всех тканях организма (интерстициальная ткань почек и канальцы, стенки вен и артерий, лёгкие, лимф.узлы, щитовидная железа, селезёнка, поджелудочная железа, головной мозг)
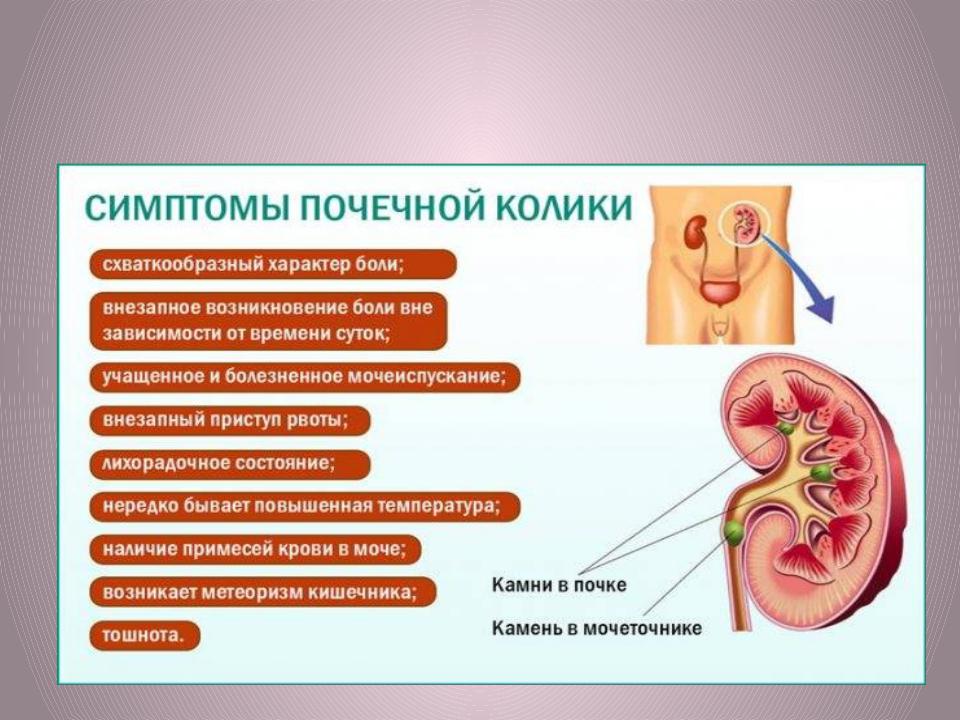
•Задержка развития, почечная колика, массивная гематурия, микробно - воспалительный процесс, повышение АД
•Повышение содержания оксалатов в крови и экскреция оксалатов с мочой 100-400 мг/сутки
•Рентген и УЗИ: картина нефролитиаза.
•Препараты магния, большие дозы витамина В6 (400 мг в день)- замедляется трансаминирование глиоксилата в глицин.
•Прогрессирующий ТИН приводящий к ХБП 5 стадии до18 лет.