

книги / Электрохимическая коррозия и защита металлов
..pdfВследствие этой особенности окислительная и восстановительная стадии химического превращения, протекающего по электрохимическому механизму, представляют собой самостоятельные электродные реакции. Их скорости зависят не только от концентрации реагирующих компонентов, но и от величины заряда металлической поверхности, т.е. от скачка потенциала на границе металла с раствором электролита. Однако эти зависимости различны: скорость окислительной (анодной) реакции увеличивается с ростом потенциала, скорость восстановительной (катодной) реакции с ростом потенциала уменьшается. В соответствии с закономерностями электрохимической кинетики:
— скорость катодной реакции
iк = K1 · СOx · exp(–αnFE/RT); (2.4)
— скорость анодной реакции
ia = K2 · CRed · exp(βnFE/RT), |
(2.5) |
где K1, K2 — константы; СOx, CRed — концентрации окислительного
ивосстановительного компонентов; n — число электронов, участвующих в процессе; F — постоянная Фарадея; Е — электродный потенциал; α и β — числа переноса, которые при условии, что переносчиком зарядов в противоположных направлениях является одна
ита же частица, удовлетворяют условию α + β = 1.
Вформе, наиболее удобной для практического использования, уравнения (2.4) и (2.5) целесообразно прологарифмировать:
ln iк = ln K1 + ln СOx – αnFE/RT.
Объединяя первые два постоянных члена ln K1 + ln СOx в новую константу А, после перевода натуральных логарифмов в десятичные
получим: |
|
lg iк = А – αnFE/2,3RT. |
(2.6) |
Введение обозначения 2,3RT/αnF = b приводит к уравнению |
|
Е = А – blgiк, |
(2.7) |
где b — характеристика скорости процесса, b–1= ∂lgiк/∂Е. Аналогичные преобразования для анодного процесса приводят
к уравнению |
|
Е = B + blgia. |
(2.8) |
|
31 |
Уравнения (2.7) и (2.8) лежат в основе закона, сформулированного Тафелем, и называются уравнениями Тафеля. Из уравнений (2.7) и (2.8) следует, что между потенциалом и логарифмом скорости электродной реакции существует линейная зависимость. Величину b соответственно называют тафелевым наклоном (имея в виду наклон линейных кривых в координатах Е – lgi). Понятие «наклон» означает, что при изменении плотности тока в 10 раз, что эквивалентно изменению логарифма этой величины на единицу, потенциал электрода, выраженный в мВ, изменяется на величину b.
Величина b является важной характеристикой и в ряде случаев служит аргументом для заключений о кинетике электрохимических реакций. Чем больше тафелев наклон, тем сильнее нужно сдвинуть потенциал электрода для увеличения плотности тока в десять раз. Поэтому величина b — характеристика степени затрудненности, торможения процесса, или, как говорят, поляризуемости электрода, на котором этот процесс протекает: чем больше b, тем сильнее поляризуется электрод.
Значение уравнений Тафеля настолько велико, что в большинстве случаев графики зависимости скорости реакции i от потенциала Е принято строить в координатах lgi — E. Тогда зависимость эта выражается прямой линией, а всякое несоответствие уравнению приводит к отклонению от прямой.
При потенциостатических измерениях Е является аргументом, а скорость реакции — функцией. Естественно было бы поэтому откладывать величину Е по оси абсцисс так, чтобы она повышалась слева направо, а плотность тока — по оси ординат. Однако в силу традиций, сложившихся еще до создания потенциостатического метода, очень часто по оси абсцисс откладывают логарифм плотности тока, а по оси ординат Е. Значение его повышается сверху вниз.
32
3. АНОДНЫЕ ПРОЦЕССЫ РАСТВОРЕНИЯ МЕТАЛЛОВ
3.1. Стадийный механизм ионизации металлов
При растворении металлов многовалентные ионы часто образуются путем последовательного отщепления электронов в не-
сколько стадий по схеме: |
|
|
|
|
— первая стадия |
Ме←→ |
K1 |
Ме+ + е; |
(3.1) |
— вторая стадия |
Ме+ ←→ |
K2 |
Ме2+ + е; |
(3.2) |
— третья стадия |
Ме2+ ←→ |
K3 |
Ме3+ + е, |
(3.3) |
где K1, K2 и K3 — константы равновесия. |
|
|||
Для двухстадийного процесса возможен такой вариант: |
|
|||
|
2(Ме← |
→ |
Ме+ + е); |
(3.4) |
|
2Ме+ ← |
→ |
Ме + Ме2+; |
(3.5) |
|
Ме← |
→ |
Ме2+ + 2е. |
(3.6) |
Исследованием стадийности реакций ионизации металлов занимались многие ученые. Теория многостадийных процессов развита в работах К. Феттера, Дж. Бокриса. В.В. Лосев экспериментально обосновал стадийный механизм растворения металлов на примере растворения индия в растворах хлорной кислоты (1955–1965 гг.). На первой стадии протекает следующий
процесс: |
|
In ← → In+ + e. |
(3.7) |
Последующее окисление этих ионов может протекать как на электроде, так и в растворе. Окисление на электроде протекает по схеме:
In+ ← |
→ |
In3+ + 2e. |
(3.8) |
Эта стадия является электрохимической, следовательно, зави- |
|||
сит от потенциала. Окисление в растворе протекает по схеме: |
|
||
In+ + 2Н+ |
→ |
In3+ + Н2 ↑ . |
(3.9) |
Скорость этого процесса зависит от концентрации кислоты и не зависит от потенциала.
33
Скорость протекания суммарного многостадийного последовательного процесса определяется скоростью самой медленной стадии. Такой медленной стадией может быть как электрохимическая, так и химическая реакция. Кинетическое уравнение в общем виде можно записать в форме:
i |
= K |
1 |
a p aq ...exp(β nFE / RT ) , |
(3.10) |
a |
|
1 2 |
|
где а1, а2… — активности компонентов раствора, принимающих участие в реакции; p, q… — показатели степени, определяющие порядок реакции по соответствующим компонентам.
Определим вид кинетического уравнения для многостадийного процесса, скорость которого лимитируется одной медленной стадией. Примем, что компоненты раствора не принимают участия в многостадийном процессе. Рассмотрим процесс, состоящий из трех одноэлектронных стадий (3.1)–(3.3). Будем считать, что все процессы протекают на поверхности электрода, а не в растворе, и поэтому их скорость зависит от потенциала.
Допустим, что медленной стадией является стадия (3.1). Ско-
рость этой стадии |
|
ia = K1 exp(β FE / RT ), |
(3.11) |
где ia — результирующая скорость процесса в токовых единицах. Прологарифмируем обе части уравнения (3.11), получим:
ln ia = ln K1 +β FE / RT . |
(3.12) |
|||||||||
Находим потенциал: |
|
|
|
|
|
|
||||
E = − |
RT |
ln K1 |
+ |
RT |
ln ia . |
(3.13) |
||||
|
|
|
||||||||
|
|
β F |
β F |
|
||||||
Подставим β = 1/2 и перейдем от натуральных к десятичным |
||||||||||
логарифмам, получим: |
|
|
|
|
|
|
||||
E = −2,3 2 |
RT |
lg K1 |
+ 2,3 2 |
RT |
lg ia , |
(3.14) |
||||
|
|
|||||||||
|
|
F |
|
|
|
F |
|
|||
где первый член есть новая константа K2 = −2,3 2 |
RT |
lg K1 , апред- |
||||
|
|
|||||
|
|
|
F |
|
||
|
|
∂ E |
|
|
||
логарифмический множитель второго члена b= |
|
|
= 120 мВ |
|||
|
∂ |
lg ia T =const |
|
|||
34 |
|
|
|
|
|
|
при постоянной температуре. Этот наклон свидетельствует о том, что для увеличения анодного тока на электроде в десять раз следует увеличить потенциал электрода на 120 мВ. Графически его находят по точкам на оси потенциалов при увеличении на порядок величины плотности тока.
Если принять, что медленной стадией является стадия (3.2), то кинетику процесса можно описать уравнением:
ia = K2 [Me+ ] exp(β FE / RT ). |
(3.15) |
Здесь концентрацию [Me+] экспериментально определить невозможно, и эту величину необходимо выразить через другие величины. Поскольку стадия (3.1) протекает быстро, то она быстро достигает равновесия, и для нахождения равновесной концентрации частиц Ме+ воспользуемся уравнением Нернста для равновесной реакции (3.1):
E = E(03.1) |
+ |
RT |
ln[Me+ ], |
(3.16) |
|
||||
отсюда |
|
F |
|
|
|
|
|
|
|
|
|
[Me+] = exp |
(E − E(03.1) )F |
. |
|
|
|
|
|
(3.17) |
||||||||||||
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
RT |
|
|
|
|
|
|
|
|
|||
Подставляем выражение (3.17) в (3.15), получим: |
|
|||||||||||||||||||||
|
|
ia= K2·exp |
(E − E(03.1) )F |
·exp |
β FE |
. |
|
(3.18) |
||||||||||||||
|
|
|
|
RT |
|
|
|
|
RT |
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Представим первую экспоненту в виде двух членов и обозна- |
||||||||||||||||||||||
чим новую константу K3 = K2 exp(−E(03.1) F / RT ), |
|
тогда |
||||||||||||||||||||
|
|
ia |
= K3 exp[(β +1)FE / RT ]. |
|
|
|
(3.19) |
|||||||||||||||
Логарифмируя выражение (3.19), находим потенциал |
||||||||||||||||||||||
|
|
E = |
2,3RT |
lg i − |
2,3RT |
lg K |
. |
|
|
|
(3.20) |
|||||||||||
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
(β + |
1) |
|
|
a |
|
(β + |
1) |
|
|
|
3 |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Получаем вновьуравнение Тафеля, нос новыми коэффициентами: |
||||||||||||||||||||||
|
2,3RT |
|
|
|
∂ E |
|
|
|
|
2,3RT |
|
|
|
2,3RT |
|
|||||||
A = − |
lg K3 |
, b= |
|
|
= |
= |
= 40 мВ. |
|||||||||||||||
|
|
|
|
|
|
|||||||||||||||||
|
(β + 1) |
|
|
∂ lg ia T |
|
|
(β + 1) |
|
|
|
|
|
3 |
|
|
|||||||
|
|
|
|
|
|
|
|
2 |
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
35 |
Если кинетику всего процесса определяет стадия (3.3), то
ia |
= K3 |
[Me2+ ] exp β FE . |
(3.21) |
|
|
RT |
|
Величину [Me2+] можно найти из уравнения Нернста для стадий
(3.1) и (3.2):
E = E(03.2) |
+ |
RT |
ln |
[Me |
2+ |
] |
, |
(3.22) |
|
||||||||
|
|
+ |
|
|||||
|
|
F [Me ] |
|
|||||
Me2+ |
|
0 |
|
||
|
|
= exp |
(E − E(3.2) )F |
, |
|
Me+ |
RT |
||||
|
|
||||
|
|
|
|||
|
|
|
0 |
|
|
|
|
[Me2+ ] = [Me+ ] exp |
(E − E(3.2) )F |
. |
|
||||
|
|
|
|||||
|
|
|
|
RT |
|
|
|
Подставляем (3.24) в уравнение (3.21), а значение [Ме+] |
|||||||
из уравнения (3.17), получим: |
|
|
|
|
|
|
|
+ |
|
(E − E(03.2 )F |
β FE |
|
|||
ia= K3·[Me |
] exp |
|
|
exp |
|
|
= |
RT |
|
|
|
||||
|
|
|
|
RT |
|
||
= K3·exp |
(E − E(03.1) )F |
|
(E − E(03.2) )F |
|
β FE |
|
|
|
|
exp |
|
|
exp |
|
= K3 |
× |
|
RT |
RT |
|
||||||
|
|
|
|
RT |
|
|
||
|
(2 + β )EF |
|
(−E(03.1) |
− E(03.2) )F |
|
|
|
|
(2 + β )EF |
|||||
× exp |
|
exp |
|
|
|
|
|
= K4 exp |
|
|
. |
|||
|
|
|
|
|
|
|||||||||
|
RT |
|
|
RT |
|
|
|
|
|
|
RT |
|||
Отсюда тафелев наклон b= |
∂ E |
= 60 |
2 |
= 24 мВ. |
||||||||||
∂ lg ia |
|
|||||||||||||
|
|
|
|
|
|
|
|
5 |
|
|
|
|
||
(3.23)
(3.24)
берем
(3.25)
Таким образом, независимо от контролирующей стадии скорость суммарногопроцессаописываетсяуравнениямитипауравненияТафеля, носразнымитафелевыминаклонамиb. Коэффициент b определяетсяне валентностью конечного продукта растворения, аконтролирующей (т.е. наиболее медленной) стадией процесса. Помере отрыва на лимитирующей стадии первого, второго, третьего электрона тафелев наклон уменьшаетсявряду120>40>25 мВ, аскоростьпроцессавозрастает.
36
3.2. Механизм растворения железа в серной кислоте (без участия растворителя и анионов растворителя)
Ионизация железа в серной кислоте выражается суммарной реакцией
Fe→Fe2+ + 2e. |
(3.26) |
||
Если бы оба электрона отщеплялись одновременно, то в соответ- |
|||
ствии с уравнением i = K |
a |
exp(β nFE RT ) |
для скорости процесса |
a |
|
|
|
(3.26) при n = 2 следовало бы записать: ia = Ka exp(2β FE 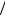 RT ) , и по-
RT ) , и по-
стоянная Тафеля ba |
= 2,3 |
|
1 |
|
|
RT |
при β = |
1 |
была бы равна 59 мВ |
|
2 |
0,5 |
|
F |
2 |
|
|||
или округленно 60 мВ. В действительности для высокочистого отожженного железа опыт дает существенно меньшее значение ba = = 40 мВ. Этот факт можно объяснить, если принять, что процесс растворения железа протекает в двестадии:
Fe ←→ |
K1 |
Fe+ + e, |
(3.27) |
Fe+ ←→ |
K2 |
Fe2+ + e. |
(3.28) |
Стадия (3.27) быстрая, следовательно, равновесная, и она предшествует медленной стадии (3.28), которая и определяет весь процесс (3.26) в целом.
Для скорости стадии (3.28) при n = 1
|
|
|
|
β F |
|
|
|
||||
iа |
= K2 aFe+ |
exp |
|
|
|
|
E . |
(3.29) |
|||
|
|
|
|||||||||
|
|
|
|
RT |
|
|
|
||||
Активность aFe+ зависит от потенциала Е по уравнению Нернста: |
|||||||||||
|
|
|
|
F |
|
|
(3.30) |
||||
|
aFe+ = K1 exp |
|
|
E |
. |
||||||
|
|
|
|||||||||
|
|
|
|
RT |
|
|
|
||||
Уравнение (3.30) подставляем в (3.29): |
|
|
|
||||||||
i |
= K exp |
|
(1 + β )F |
E |
, |
(3.31) |
|||||
|
|||||||||||
а |
|
|
|
|
|
|
|
|
|
|
|
|
|
RT |
|
|
|
||||||
где K = K1 · K2.
37

Отсюда
b = (∂ E /∂ |
lg i |
) = 2,3 |
|
1 |
|
RT= 40 мВ. |
a |
а |
T |
(1 |
+ β ) |
|
F |
|
|
|
|
Таким образом, стадийный характер ионизации, который здесь сводится к наличию предшествующей равновесной электрохимической стадии, приводит к уменьшению постоянной Тафеля, т.е. усилению влияния потенциала на скорость реакции.
3.3. Анодное растворение металлов с участием компонентов раствора
Изложенные в подразделах 3.1 и 3.2 вопросы механизма коррозионных процессов относились к случаям, когда скорости собственно анодных реакций растворения металлов не зависели от состава раствора. В действительности же нередко на скорости процессов растворения влияет не только потенциал, но и концентрации компонентов раствора, чаще всего анионов электролита. Это влияние можно объяснить на основе развитого Я.М. Колотыркиным учения, согласно которому электрохимические реакции ионизации атомов металла, как правило, включают стадии химического или адсорбционно-химического взаимодействия поверхностных атомов металла с компонентами среды. Такое взаимодействие приводит к образованию устойчивых или промежуточных комплексов металла с компонентами раствора непосредственно в электрохимической стадии. При хемосорбции компонента, участвующего в реакции растворения металла, реализуются определенная прочность связи между адсорбированной частицей и электродом и определенная степень заполнения поверхности. Эта степень заполнения возрастает по мере смещения потенциала в положительном направлении иопределяет скорость растворения металла.
В качестве участников реакции при растворении металлов могут выступать различные анионы, а также молекулы растворителя. При одновременном нахождении в растворе нескольких компонентов различной природы возможно их участие либо в параллельных, либо в последовательных стадиях.
38
При параллельном протекании двух реакций растворения металла с участием анионов An1− и An2− :
Ме + p An1− → |
Me(An1 )(pn1 − p )+ + n1e, |
(3.32) |
Me + qAn2− → |
Me(An2 )(qn2 −q )+ n2e, |
(3.33) |
где p и q — коэффициенты, эти анионы могут в одних случаях не мешать друг другу (при малых степенях заполнения Θ). При этом каждая из реакций будет протекать на всей поверхности электрода. В других условиях может иметь место конкурирующая адсорбция указанных анионов, при которой идет их «борьба» за место на электроде и соответственно за участие в электродной реакции. В первом случае скорость растворения металла выражается уравнением:
|
ia |
= K1aAnp exp(β |
1n1FE / RT )+ K2aAnq |
exp(β |
2n2 FE / RT ) , (3.34) |
|
|
1 |
|
2 |
|
где aAnp |
, aAnp — активностьанионов, β1 иβ2 — коэффициентыпереноса. |
||||
1 |
|
2 |
|
|
|
Из (3.34) следует, что при определенных соотношениях между активностями анионов An1− и An2− одним из слагаемых в выражении
ia можно пренебречь. В этих условиях суммарная скорость процесса практически определяется скоростью одной из реакций (3.32) или (3.33). При «промежуточных» значениях активностей анионов An1−
и An2− и потенциалов, когда оба слагаемых в уравнении (3.34) соизмеримы, зависимость тока от активностей анионов An1− и An2− и от
потенциала отличается от той, которая имеет место в растворах, содержащих ионы только одного вида.
Указанный механизм независимого, параллельного протекания двух реакций реализуется сравнительно редко. Чаще же адсорбция одного из анионов приводит к изменению условий и закономерностей адсорбции другого аниона. Например, возможно выполнение уравнения:
ia = K1Θ 1 exp(β 1n1FE / RT ) + K2Θ 2 exp(β 2n2 FE / RT ), (3.35)
где Θ1 и Θ2 — степени заполнения поверхности электрода анионами
An1− и An2− .
39
Взаимное влияние частиц, адсорбирующихся на металле при его растворении, может иметь и иной характер. Адсорбция одного из компонентов может способствовать адсорбции другого компонента с образованием комплекса, включающего обе адсорбированные частицы.
В результате конкурирующей адсорбции двух находящихся
врастворе частиц — участников реакции растворения металла одна из них может вызвать замедленное растворение. Это может быть
втех случаях, когда частица An1− , растворение с участием которой
происходит медленнее, чем с частицей An2− , обладает большей адсорбционной способностью. Вытесняя с поверхности кинетически более активный компонент An2− , частица An1− будет тормозить рас-
творение. По такому механизму замедляется, например, растворение железа в серной кислоте под действием добавок ионов хлора.
Адсорбированные на металле частицы могут тормозить растворение и в отсутствие в растворе второй адсорбирующей частицы. По теории Я.М. Колотыркина, функция адсорбирующегося компонента (стимулирование или замедление коррозии) определяется соотношением прочностей связей адсорбированного компонента с металлом и с сольватирующими его молекулами. Для проявления частицей ускоряющего действия на растворение металла она должна быть достаточно прочно связана и с металлом и с раствором. Если связь с сольватной оболочкой относительно слаба, а с металлом очень прочна, частица утрачивает связь с раствором, остается на поверхности металла и замедляет его растворение.
При оценке роли компонентов среды в процессе растворения металла необходимо принимать во внимание, что его поверхность обычно энергетически неоднородна, т.е. адсорбция даже одной и той же частицы на одних участках поверхности может быть стимулирующей, а на других — ингибирующей (замедляющей коррозию). То же относится и к частицам разной природы при их совместном нахождении в растворе. Существенно при этом, что прочность связи адсорбированной частицы с металлом, а следовательно,
40