

Лабораторная работа
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
Целью работы является ознакомление студентов с основными положениями теории валентных связей для объяснения образования и строения комплексных соединений, с их номенклатурой.
Программа коллоквиума
1. Электронные конфигурации элементов d - ряда.
а)Какие орбитали и электроны d - элементов являются валентными ? Чем это объясняется?
б)Почему для d - элементов характерно разнообразие степеней окисления? Как изменяется в ряду d- элементов высшая степень окисления элементов ?
2.Электронные конфигурации ионов, отвечающие различным степеням
окисления d- элементов,
II. Комплексные соединения.
1. Основные положения теории валентных связей для объяснения
образования и строения комплексных соединений.
а) Объяснить особую склонность d- элементов к образованию комплексных соединений, тип гибридизации.
б) Комплексообразователь, лиганды, координационное число, устойчивость комплексных соединений, константа нестойкости. Магнитные свойства комплексных соединений, образование соединений с комплексным катионом и комплексным анионом.
2. Номенклатура комплексных соединений.
3. Получение и свойства комплексных соединений.
а) Получение комплексных соединений с помощью реакции замещения лигандов во внутренней сфере.
б) Получение комплексных соединений с помощью окислительно-. восстановительных реакций.
в)Установление координационной формулы комплексной соли по реакциям, характерным для ионов внешней сферы.
Основополагающие представления о комплексных соединениях ввел в науку Альфред Вернер (1898 г.). В развитии химии комплексных соединений большую роль сыграли труды Л.А.Чугаева и его учеников.
В определении понятия "комплексное соединение" нет полного единства. Это обусловлено разнообразием комплексных соединений и разнообразной гаммой их характерных свойств. Комплексными называются соединения, в узлах кристаллов которых находится комплексы, способные к самостоятельному существованию в растворе. Следует подчеркнуть, что такое определение, конечно, далеко не исчерпывающее и оно применимо лишь в известных пределах.
Согласно Вернеру, в большинстве комплексных соединений различают внутреннюю и внешнюю сферы. Например, в комплексных: соединения К2[BeF4] и [Zn(NH3)4] Cl2 внутреннюю сферу составляют группировки атомов (комплексы) [BeF4]-2 и [Zn(NH3)4]+2 ,а внешнюю сферу - соответственно К+ и С1-, Центральный атом или ион внутренней сферы называется комплексообразователем, координированные вокруг него молекулы или ионы противоположного знака наминаются лигандами (адцендами). В формулах комплексных соединений внутреннюю сферу (комплекс) часто заключают в квадратные скобки. Число, показывающее, сколько лигандов удерживает комплексообразователь, называется координационным. Чаще всего оно равно 6 или 4, реже -2.
Координационные числа зависят от объемов комплексообразоваятеля и лигандов и их зарядов.
Чем больше объем комплексообразователя и меньше объем лигандов, тем больше лигандов может разместиться вокруг комплексообразователя и наоборот. Так, Al+3 может удержать шесть ионов F - с малым радиусом: Na [AIF6] и только четыре более объемистых лиганда ( Вг-1 или J-)
Увеличение заряда комплексообразователя и понижение заряда лигандов способствует повышению координационного числа комплексообразователя. Так, у Сu+ и Аu+ координационные числа 2, а у Cu+2 и Аu+2 координационные числа 4.
Мерой устойчивости комплекса служит ее константа диссоциации, которая в этом случае называется константой нестойкости комплекса и обозначается через Кн.
(MeLn)+m Me+m + nL
Кн
=



где Me - металл, L - лиганд.

Диссоциация комплексных ионов обычно незначительна и величина Кн - мала. Очевидно, что чем больше константа нестойкости, тем менее устойчиво комплексное соединение.
Так, среди однотипных соединений
[Aq(NO2)2]-1 [Ag(S203)2)]-3 [Ag(NH3)2]+1 [Ag(CN)2]-1
KH 1,3*10-3 1*10-13 6,8*10-8 1*10-21
Устойчивость комплекса возрастает при переходе от [Ag(NO2)2]-[Ag(CN)2]
Заряд комплексного иона равен алгебраической сумме зарядов комплексообразователя и лигандов или заряду ионов, находящихся во внешней сфере, взятых с противоположным знаком. Например:
внешняя сфера K2[BeF4] внутренняя сфера
где ион Be+2- комплекеообразователь, лиганды- кислотные остатки фтористоводородной кислоты Координационное число комплексов образователя равно 4, суммарный заряд лигандов равен -4„следовательно , заряд комплексного иона +2 - 4 = -2. Зная заряд комплексного иона и заряд лигандов, можно определить валентность комплексообразователя. Например, в соединении состава K3[Fe(CN)6] заряд комплексного иона равен -3 (K3 [Fe(CN)6 ]-3. Тогда, обозначив валентность комплексообразователя через X, находим: X +(-.6) = - 3, X = +3, CN-- кислотный остаток цианистоводородной кислоты, координационное число равно 6.
По характеру электрического заряда различают катионные, анионные и нейтральные комплексы. Катионный комплекс можно рассматривать, как образованный в результате координации вокруг положительного иона нейтральных молекул.
Рассмотрим процесс образования комплексного соединения, в котором роль лигандов играют нейтральные молекулы аммиака NН3 , а центральным атомом является положительный ион какого-либо металла, например, меди, взятой в виде хлорида или другой соли. Молекулы аммиака, имеющие по одной не поделенной паре электронов, являются донорами, а ион меди - акцептором.
При взаимодействии возникают четыре донорно-акцепторные связи, и получается комплексное соединение, относящееся к амминам:
H̤ +2
H:N̤̤̤:H
□ H̤ H̤ H̤
□ Cu+2 □ + H :N̤: = H :N̤ : Cṳ : N̤: H = [Cu(NH3)4]Cl2
□ H H H
H:N̤:H
H
Аналогичным образом можно показать, что при взаимодействии иона Cr+3 с шестью молекулами воды образуется комплексный ион
□ □ □ ОН2
□ Cr+3 □ + 6: ОН2 → ОН2 Cr ОН2
□ ОН2 ОН2 ОН2
В этом случае, акцептором является центральный атом - хром, а донорами электронной пары - молекулы воды.
В анионном комплексе роль комплексообразователя играет атом положительной степени окисления, а лигандами являются атомы отрицательной степени окисления.
Нейтральные комплексы образуются при координации вокруг атома молекул , а также при одновременной координации вокруг положительного иона - комплексообразователя отрицательных ионов и молекул: [Fe(С0)5] , [Pt(NH3)2Cl2]
Электронейтральные комплексы, следовательно, являются комплексными соединениями без внешней сферы.
Роль комплексообразователя может играть любой элемент периодической системы. В соответствии со своей химической природой неметаллические элементы обычно дают анионные комплексы, в которых роль лигандов играют атомы наиболее электроотрицательных элементов, например: К[PF6] , К3[Р04] .
Что же касается типичных металлических элементов, то способность к образованию комплексных соединений у них выражена слабо. Имеющиеся немногочисленные комплексные ионы являются катионами, например: Ga(NH3)8Cl2 ,[S(H2 O)6]Cl2.
Склонность к комплексообразованию сильно выражена у элементов, относящихся к переходной группе периодической системы. Это обусловлено наличием свободных орбиталей.
При комплексообразовании путем присоединения лигандов, электроны комплексообразующего атома металла спариваются, оставляя отдельные энергетические ячейки, которые могут быть заняты свободными электронными парами координированных групп лигандов.
В процессе комплексообразования у элементов IV периода обнаруживается тенденция к созданию электронной конфигурации, сходной с электронной конфигурацией инертного газа криптона, завершающего этот период. Его электронная конфигурация может быть записана следующим образом: Is22s 22p63S2ЗрбЗd104s24p6 Атом железа с порядковым номером 26" должен иметь"" конфигурацию
Is22s22p63s23p63d64s2 Для заполнения электронами своей оболочки до оболочки криптона иону Fe+3 недостает 12 электронов, из которых четыре заполняют орбитали Зd-слоя до Зd 10 ,два -орбиталь 4s и шесть- 4р -орбитали. Следовательно, ион Fer'3 может явиться акцептором 6 электронных пар, и его координационное число должно равняться 6.
Номенклатура комплексных соединений
В 1958 году на Менделеевском съезде теоретической и прикладной химии была принята новая номенклатура комплексных соединений.
Название комплексного катиона начинают с указания внутренней сферы, причем в, следующей последовательности: лиганды (если они смешанные, то сначала нейтральные молекулы, а затем- анионы)„Если одинаковых лигандов во внутренней сфере комплекса больше одного, то их количество отмечают при помощи греческих числительных (2-ди , 3-три, 4-тетра, 5„цента ,6-гекса, 7-гептаs 8-окта). Ниже перечислены наиболее часто встречающиеся лиганды;
F- - фторо- S-2 тио- ОН- чпидроксо—
С1-. хлоро- СО-23- карбонатов- NH3 аммин-
Вr--бромо- CN-- циано- H2O акво
I- -иодо- CNS- родано- СО -карбонил-.
NO-2 нитро- PO-34 фосфато
NO-3-нитрато- NH2 амидо-
После названия лиганда называют центральный ион (ион-комплексообразователь); при этом в основу кладут русские незнания элементов в родительном падеже, а затем - латинские названия внешней .сферы.
[Со(NH3)4С03]С1 -тетраамин-карбонато-кобальта (Ш)xлорид,
[Al(H 2O)6] CI3 - гексаакваалюминия хлорид
Соль содержит комплексный анион
Вначале также называют лиганды, начиная с нейтральных молекул с введением в наименование (если это нужно) греческих числительных» После этого называют комплексообразователь, используя латинское название элемента с прибавлением суффикса - ат. Студень окисления центрального иона (если это необходимо) также отмечается римскими цифрами в скобках. Наконец, называют катион, образующий внешнюю сферу комплексного соединения (используется русское название элемента в родительном падеже)
K[CO(NH3)2 (NO2)4] - диамминтетранитрокобальтат калия
K3 [Fe(CN)6] - гексацианоферрат (Ш) калия
K2[PtCl6]- гексахлороплатинат (IУ) калия
Наименование нейтральных комплексов составляется из названий лигандов (в последовательности, указанной для комплексных, катионов) и русского названия центрального элемента в именительном падеже. При этом валентность комплексообразователя в названии не указывается,
[Pt(NH3)2Cl4] - диамминтетрахлороплатина
[Ni(CO)4]- тетракарбрнилникель
[CO(NH3)3(NO3)3] триамминтринитрокобальт
Для объяснения образования и свойств комплексных соединений в настоящее время применяется метод валентных связей ( ВС)теория кристаллического поля (ТКП) и метод молекулярных орбиталей (МО).
Метод валентных Связей. Согласно этому методу образование комплексов осуществляется за счет донорно-акцепторного взаимодействия чаще всего неподеленных электронных пар лигандов и свободных орбиталей комплексообралопателя.
Р азберем
случай, когда координационное число
комплексообразователя равно 4. Так,
двухзарядный катион
Zn+2
, отдав два 4s -электрона,
имеет на внешнем уровне Зd10электронов
азберем
случай, когда координационное число
комплексообразователя равно 4. Так,
двухзарядный катион
Zn+2
, отдав два 4s -электрона,
имеет на внешнем уровне Зd10электронов
Для достижения электронной конфигурации криптона катион должен принять.8 электронов. Если роль донора играют четыре нейтральные молекулы аммиака, которые и дают эти восемь электронов, то образующийся комплекс несет два положительных заряда.
Рассмотрим процесс образования этого комплексного соединения. Здесь центральным атомом является положительный ион металла,Zn+2 взятый в виде растворимой соли. Молекулы аммиака, имеющие по одной неподеленной паре электронов, являются, донорами, а ион цинка - акцептором. При взаимодействии возникают четыре донорно-акцепторные связи за счет четырех свободных орбиталей иона Zn+2 и четырех молекул аммиака, представляющих каждый по одной неподеленной паре электронов и заполняющих 4 орбитали.
Получается комплексное соединение:

Ион Fe+2 играет роль акцептора и представлявет для образования связи 6 орбиталей с d2sр3 -гибридизацией, Роль лигандов играют ионы CN- они имеют по одной неподеленной паре электронов атома азота и углерода и являются донорами. При взаимодействии возникают шесть донорно-акцепторных связей:

Октаэдрическая формула молекулы объясняется d2sp3 гибридизацией.
Теория кристаллического поля
В 1930 году Ван-Флек и Бэте выдвинули теорию кристаллического поля, которая получила распространение среди химиков гораздо позже (1951 г.), когда на ее основе удалось истолковать спектры поглощения комплексных соединений.
Теория кристаллического поля основана на предположении, кто между комплексообразователем и лигандом осуществляется чисто электростатическое взаимодействие (ионная связь). В теории кристаллического поля учитывается влияние электростатического поля лигандов на энергетическое состояние электронов комплексообразователя.
Рассмотрим основные положения теории кристаллического поля на примере одноядерных комплексов d-элементов.
Напомним, что пять d-орбиталей по-разному располагайся в пространстве относительно атомного ядра.

Орбитали dx и d xy обычно обозначают d j а орбитали (dx и dyz обозначают de . В свободном атоме или ионе все пять d- орбиталей одного и того же подуровня имеют одинаковую энергию, то есть они вырождены. Следует отметить, что d j орбитали, направленные вдоль осей, участвуют в образовании € -связей, а d j орбитали, локализованные между осями, являются несвязывающими или участвуют в образовании P связей.
Рассмотрим типичный октаэдрический комплекс ML6 где L - лиганда. В нем электрическое поле, создаваемое неподеленными 6~ парами шести лигандов, которые, окружают ион Мп+, направлено вдоль осей х, у и z. :

о - центральный атом • - лиганда
Представим себе, что свободный ион по
мере приближения к нему лигандов
постепенно начинает испытывать влияние
поля. Пока лиганды находятся далеко от
металла, можно считать, что это поле
сферически симметрично и под его
воздействием энергия всех электронов
постепенно увеличивается. Но по мере
приближения лигандов становится заметно,
что их поле направлено вдоль осей и на
некоторые электроны действует
сильнее, чем на другие. Вдоль осей
направлены d j-орбитали,
поэтому находящиеся на них электроны
будут более сильно отталкиваться
полем неподеленных пар лигандов, чем
электроны, находящиеся на de-орбиталях.
Под влиянием поля октаэдрически
расположенных лигандов п ять
вырожденных сгорбит а- лей распадутся
на две группы: высокоэнергетический
дублет и низкоэнергетический триплет
de.
ять
вырожденных сгорбит а- лей распадутся
на две группы: высокоэнергетический
дублет и низкоэнергетический триплет
de.
Разница энергий верхнего дублета и нижнего триплета обозначается ∆.
Значение параметра расщепления ∆ изменяется от комплекса к комплексу и зависит от природы лигандов и иона металла, Поле, создаваемое лигандами, зависит от их природы и особенно от легкости, с которой электроны лиганда возмущаются ионом. На практике лиганды можно расположить в порядке уменьшения влияния их кристаллического поля:
C N ~ > NH3 > H2O > F- > CI- > Br >I
Этот порядок по существу не зависит от иона металла, к которому они присоединены.
Может показаться, что этот ряд, часто называемый спектро-химическим рядом, вначале несколько необычен, так как можно было бы ожидать, что F- будет создавать большее поле, чем CN- Однако нужно помнить, что электроны иона фтора крепче удерживаются ядром и не легко возмущаются ионом металла, как электроны более "мягкого" цианид-иона.
На величину расщепления уровней энергии кристаллическим полем влияет степень окисления центрального атома и тип имеющихся у него d-электронов. С увеличением степени окисления d- элемента с возрастания заряда иона) увеличивается, так как лиганды ближе подходят к центральному иону и, следовательно, вызывают большее расщепление d-уровня. В подгруппах d-элементов при переходе от четвертого к пятому и в особенности к шестому периоду ∆ однотипных комплексов заметно возрастает. Это объясняется тем, что 4d 5d орбитали простираются в пространстве дальше от ядра, чем 3-орбитали. Это отвечает более сильному отталкиванию электронов и лигандов и соответственно большему расщеплению 4 d- и 5 d-уровней по сравнению с 3d- уровнем.
Магнитная восприимчивость. Как известно, атомы, содержащие неспаренные электроны, парамагнитны. В сущности неспаренный электрон ведет себя как маленький магнит, то есть "втягивается" в приложенное поле. Если на орбитали два электрона, их спины должны быть противоположно направлены, и их равные противоположно направленные магнитные моменты будут компенсировать друг друга. Когда все орбитали имеют по два электрона, атом диамагнитен. Атом с неспаренным электроном парамагнитен. Магнитный момент Mm, связанный с неспаренным электроном, является векторной величиной, он может быть измерен на опыте, а также вычислен по формуле:
Mm =  где n-число неспаренных
электронов
где n-число неспаренных
электронов
Теория кристаллического поля достаточно просто и наглядно объясняет магнитные свойства комплексов, их спектры и ряд других свойств. Для понимания этих свойств необходимо знать характер распределения электронов по d-орбиталям иона, находящегося в поле лигандов. Последнее зависит от соотношения величины энергии расщепления и энергии отталкивания электронов друг от друга.
Когда поле лигандов велико, велик параметр расщепления ∆ электроны сначала полностью заполняют орбитали с меньшей энергией, а уж затем орбитали с большей энергией.
Когда поле лигандов мало, будет мал ∆ пять d-орбиталей последовательно заполняются сначала по одному, а затем по второму электрону.
В качестве примера рассмотрим характер распределения 3 d электронов иона Со+3 при образовании октаэдрических комплексов [CoF6]3-
и [Co(NH3)6]3+
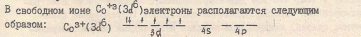
Рассчитано, что энергия отталкивания электронов одной и той же орбитали для иона Со+3равна 251 кДж/моль, энергия расщепления ∆1 его 3d орбиталей в октаэдрическом поле ионов составляет 156 кДж/моль, а в поле молекул H3N ∆2 = 265 кДж/моль.
Таким образом, в поле иона F- значение ∆ невелико, поэтому число непарных электронов на орбиталях расщепленных уровней Со3+такое же, как в свободном ионе. Но в поле, создаваемом молекулами H3N, энергия расщепления большая и энергетически более выгодно, когда d -электроны иона Со3+располагаются только на нижних -d-орбиталях.

По характеру распределения электронов по орбиталям Со3+ ион [CoF6]3- является высокоспиновым (четыре непарных электрона), а ион [Co(NH3)6]3+ низкоспиновым комплексом (непарные электроны отсутствуют).
В соответствии с характером распределения электронов по d-орбиталям комплекс [Co(NH3)6]3+ диамагнитен, а комплекс[СоF6]3+„ парамагнитен:

Величину параметра расщепления обычно определяют по спектрам поглощения соединений. Кванты света, возбуждающие переход электронов с нижних d - орбиталей на верхние, соответствуют видимой области спектра, и значения ∆ лежат в пределах 1 эв<∆<4 эв. Этим объясняется тот факт, что соединения d-элементов обычно окрашены.
Один из недостатков простой теории кристаллического поля состоит в том, что все действующие силы считаются чисто электростатическими. Перераспределение электронов (образование ковалентных связей) между ионом металла и лигандами не рассматривается, за исключением случая, когда оно возникает из-за поляризации неподеленных пар электронов ионом металла.
Экспериментальная часть
1.Ионы двойной соли
В три пробирки налить по 2 мл раствора KAI(SO4)2, в одну добавить раствор кислого виннокислого натрия Na 4C 4H 4O 6, в другую - несколько капель раствора NaOH, в третью - раствор соли бария BaCl2. Составить ионные уравнения всех трех реакций. На присутствие каких ионов в исследуемом растворе указывают эти реакции? Составить уравнение электролитической диссоциации.
2 Ионы комплексной соли
В пробирку налить немного раствора хлорного железа и прибавить раствор роданистого калия KCNS . Что образуется? Составьте уравнения реакций. На наличие какого иона в растворе указывает эта реакция?
Провести аналогичную реакцию, взяв вместо раствора FeCI3, раствор красной кровяной соли К3 [Fe(CN)6] , Появляется ли красное окрашивание? Есть ли в растворе красной кровяной соли ионы
трехвалентного железа Fe4. Составить уравнение электролитической диссоциации.
3. Комплексные соединения в реакциях обмена
а) К разбавленному подкисленному раствору FeCI3 добавить разный объем раствора желтой кровяной соли К3 [Fe(CN)6]. Что образуется? Составить уравнение реакции.
б) К свежеприготовленному раствору FeSO4 прибавить растворы красной и желтой кровяных солей. С какой из солей получается си¬нее окрашивание? Написать уравнение реакции в молекулярной и ионной формах.
в) К раствору CuSO4 прибавить раствор желтой кровяной соли. Что образуется? Написать уравнение реакции.
г) К раствору KCI прибавить раствор комплексной соли Na2[Co(NO2)6] . Выделяется желтый кристаллический осадок
KNa2[Co(NO2)6]. Составить уравнение реакции.
4, Образование аммиакатов меди и цинка
Налейте в пробирки 2 мл раствора CuSO4 и 2 мл ZnSO4, подействуйте на него раствором NaOH. Составьте уравнение реакции и отметьте цвет осадка. Понемногу добавляйте в пробирку концентри-рованного раствора аммиака. Наблюдайте за растворением осадка и изменением окраски вследствие образования ионов [Cu(NH3)4]+2 и [Zn(NH3)4]+2 Составьте уравнение реакции.
5.Соединения с комплексным отрицательным ионом.
а) На часовой стекло поместить по 3-4 капли концентрированных растворов хлористого кобальта и роданистого аммония Раствор окрашивается в синий цвет вследствие образования (NH4)2[Сo(CNS)4].
Составьте уравнение реакции.
б) Налейте в пробирку немного раствора Hg(NO3)2 и несколько капель раствора KI. Наблюдайте образование осадка.
Напишите уравнение реакции.
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
Курс общей химии предполагает знакомство студентов с некоторыми основными положениями, химической термодинамики. Между тем, в современных учебниках по общей химии данный материал изложен явно недостаточно. В то же время студент 1 курса встречает существенные трудности при самостоятельном изучении учебных пособий по химической термодинамике, содержащих к тому же вопросы, не предусмотренные программой для технических вузов.
Настоящий раздел призван помочь студентам получить общие представления о ряде наиболее важных закономерностей химической термодинамики и на этой основе научить их анализировать простые химические реакции.
1. Основные понятия химической термодинамики. Термодинамическая система и термодинамический процесс. Внутренняя энергия, теплота, работа.
2. Первый закон термодинамики. Адиабатный, изотермный, изохорный и изобарный процессы. Энтальпия. Стандартная энтальпия образования. Энтальпия химической реакции.
3. Термохимия. Основные законы термохимии. Термохимические расчеты.
4. Термодинамическая вероятность и энтропия.
5. Второй закон термодинамики. Энергия Гиббса. Стандартный изобарный потенциал образования. Направление процесса, энтальпийный и энтропийный факторы.
6. Константа химического равновесия. Связь константы химического равновесия с изобарном потенциалом, энтальпией и энтропией процесса.
7. Изобарный потенциал и направленность окислительно-восста-новительных реакций.
1 Основные понятия химической термодинамики
Химическая термодинамика является одним из разделов физической химии. Химическая термодинамика изучает различные химические и физико-химические процессы путем исследования изменений энергетических состояний участвующих тел. Термодинамической системой называют тело или группу взаимодействующих тел. мысленно выделяемых в пространстве. Остальная часть пространства называется окружающей средой.
Система считается изолированной, если она лишена возможности обмениваться с окружающей средой массой и энергией. Система, которая обменивается с Окружающей средой только энергией, называется замкнутой.
Системы разделяются на два больших класса - гомогенные и гетерогенные. Гомогенной называется физически однородная система имеющая одинаковые физические свойства во всех своих частях, например, смеси различных: газов, растворы как жидкие, так и твердые Гетерогенной называется система, состоящая из нескольких однородных частей с разными физическими свойствами.
Система всегда находится в том или ином состоянии, которое характеризуется всей совокупностью ее физических и химических свойств: объемом V , давлением Р, температурой Т, плотностью d и т.д. Такие свойства называются функциями состояния системы. При некоторых определенных сочетаниях значений функций состояния система находится в состоянии равновесия, то есть в таком состоянии, которое остается неизменным во времени при отсутствии изменений окружающей среды.
Свойства неизолированной системы могут изменяться, при этом естественно изменяется и состояние системы, что и представляет собой термодинамический процесс. Если в результате процесса система через ряд промежуточных состояний возвращается к исходному состоянию, то такой процесс называется циклом.
2. Первый закон термодинамики в применении к химическим реакциям.
Неотъемлемым свойством любой системы является энергия, которая представляет собой общую меру различных форм движения материи. Основным термодинамическим понятием является внутренняя энергия системы U, которую рассматривают как совокупность всех видов энергии, связанных со всевозможными движениями и взаимодействиями внутри системы. Это энергия молекул и энергия электронов, энергия взаимодействия атомов внутри молекулы, энергия межмолекулярного взаимодействия и т.д. Запас внутренней энергии зависит только от состояния системы, то есть внутренняя энергия является функцией состояния. Это означает, что изменение внутренней энергии .не зависит от характера термодинамического процесса, а
определяется разностью внутренних энергий системы в конечном U- и начальным U1 состояниях: U = U2 – U1
В состав внутренней энергии не входят потенциальная и кинетическая энергии системы в целом, поскольку Они зависят не только от внутренних свойств самой системы, но и от свойств тел окружающей среды.
Энергию взаимодействия атомов в молекуле называют химической энергией. Изменение этой энергии имеет место при химических превращениях и обусловлено в первую очередь различиями в энергии разорванных и образованных химических связей в процессе реакции.
Величина энергии отдельной химической связи очень мала. Например, чтобы разорвать связь в одной молекуле водорода достаточно потратить энергию в количестве 7,22 –10-19 Дж. Однако химическая термодинамика имеет дело с системами, содержащими большое число частиц, а, следовательно, и с достаточно заметными количествами энергии. Так, энергия химической связи, приходящаяся на I моль водорода (2г), составляет 4,35 *105 Дж.
Совершая какой-либо процесс, система может обмениваться с окружающей средой энергией как в форме теплоты Q, так и в форме работы А. (Первый закон термодинамики (является частным случаем закона сохранения энергии ) утверждает, что убыль внутренней энергии системы расходуется на выделение теплоты и совершение
работы: -∆U= - Q+А (2)
В отличии от внутренней энергии теплота и работа зависят от способа осуществления процесса и не является функциями состояния (нельзя сказать, что данная система характеризуется определенной теплотой или работой). Теплота или тепловой эффект процесса - это количественная характеристика энергии, которую система в ходе процесса получила от окружающей среды (отдала окружающей среде) в форме кинетической энергии теплового, то есть хаотического движения частиц(атомов, молекул, электронов и т.п.). Работа характеризует обмен энергией в форме кинетической энергии направленного, упорядоченного движения частиц.
Тепловым эффектом сопровождается подавляющее большинство химических реакций. Реакции, протекающие с выделением тепла, называются экзотермическими, а с поглощением – эндотермическими .Обычно тепловой эффект относится к 1 молю образовавшегося или сгоревшего вещества и измеряют в килоджоулях (кДж).
Под работой в химических процессах подразумевается в основном работа против сил внешнего давления р. В первом приближении она выражается так: А = р∆V , (3)
где V = V2 – V1 -разность объемов продуктов реакции и объемов исходных: веществ V1 .
При разных процессах перехода из состояния 1 в состояние 2 система обменивается с окружающей средой различными количествами работы и теплоты. Но независимо от характера процесса разность между теплотой и работой остается величиной постоянной и равна изменению внутренней энергии:
Qi - Ai = U2 – U1 = const. (4)
В применении к процессам, ограниченным определенными условиями, уравнение (2) упрощается и процесс можно охарактеризовать одной из величин, входящих в уравнение (2):
1. При адиабатном процессе, то есть процессе в случае изолированной системы Q = 0. Однако система может совершать работу за счет внутренней энергии, например, расширение нагретого газа, связанное с его охлаждением:
Q=∆U + А = 0, А =-∆U (5)
2. При изотермном процессе Т = const Для системы из идеальных газов ∆U = 0, так как в этом случае запас внутренней энергии зависит только от температуры. Все тепло, получаемое системой, переходит в работу:
QT= А . (6)
3. Пря изохорном процессе V-const работа расширения невозможна А = 0. Тоща при условии отсутствия других форм работы вся поглощенная теплота расходуется на увеличение внутренней энергии
QV= ∆U. (7)
В этом случае теплота процесса равна изменению термодинамической функции состояния U , Это важно при исследовании химических реагирующих систем. Так, экспериментальное определение теплоты реакции горения обычно осуществляют при постоянном объеме.
4. При изобарном процессе Р = const, Так как большинство химических реакций проводят при постоянном давлении, данное условие наиболее часто встречается в химии. При этом процесс связан с изменением объема и совершением работы расширения (или сжатия)
А = p(-V2-V1) и изменением внутренней энергии:
Q = (-U2 –U1) + p(V2 – V1) = (U2+PV2) - (U1+ pV1) (8)
Так как и p и V являются функциями состояния (или свойствами) системы, сумма U+pV также является свойством системы и обозначается через Н: Н = U + pV. (9)
Н имеет размерность энергии и называется энтальпией, которая складывается из внутренней энергии pV «объемной энергии» pV. Абсолютное значение энтальпии, также как и внутренней энергии, знать невозможно. Однако, разность энтальпии в двух состояниях может быть найдена. Для изобарного процесса эта разность равна тепловому эффекту, в чем и проявляется наиболее ясно физический смысл энтальпии:
Qp = H2 - H1 = ∆Н, (10)
где Н - изменение энтальпии или просто энтальпия химической реакции.
Для экзотермических реакций энтальпия самих молекул понижается, и изменение энтальпии реакции имеет отрицательный знак (∆Н < 0). В случае эндотермических реакций изменение энтальпии положительно (∆Н > 0). Таким образом, энтальпия реакции равна по величине и противоположна по знаку тепловому эффекту реакции при постоянном давлении. Например:
∆Н = - 100 кДж/моль
СН4 + С12 — CH3CI + HCI Qp = 100 кДж/моль
Энтальпия реакции может быть представлена как разность энергии диссоциации разрывающихся и образующихся связей. Для экзотермических реакций энергия образующихся связей превышает энергию разрывающихся связей:
C H3
– H + Cl – Cl (670) CH3
– Cl + H – Cl (770)
H3
– H + Cl – Cl (670) CH3
– Cl + H – Cl (770)
H=670-770=100 (к/Дж/моль)
В случае эндотермических реакций образующиеся связи, наоборот, должны быть менее прочны. В результате такие реакции протекают частично, либо совсем не реализуются, например:
C H3 – H + I – I (578) CH3 – I + H – I (519)
∆H=578-519=59 (к/Дж/моль)
Поскольку энтальпия зависит от температуры (в меньшей степени и от давления), сопоставлять можно только энтальпии реакций, отнесенные к одинаковым условиям. В качестве последних принимаются так называемые стандартные состояния веществ, то есть их наиболее стабильные формы при Т = 298,15°К и р = 101 325 Па (1атм), например, йод - твердый, углерод-графит, кислород - газообразный, вода - жидкая (хотя, иногда применяют в качестве стандартного и парообразное состояние воды) и т.д. Например, в реакции:
C(граф.) + O2(г) → CO2 (г). ∆H˚298 = 393,5 кДк/моль (1)
стандартным состоянием углерода является графит (наиболее стабильная форма при 298°К), а кислород и двуокись углерода находятся при давлении 1 атм. Символ H˚298 означает стандартную энтальпию реакции, отнесенную к 1 молю продукта.
Запись химических реакций с указанием теплового эффекта (или энтальпии) называют термохимическим уравнением. При этом также указывается агрегатное состояние принимающих участие в реакции веществ: (к) - кристаллическое, (ж) - жидкое, (г) - газообразное, (р) - раствор, например:
H 2(Г) + 1/2 O 2(г) H2O(ж) ∆H˚298 = - 286,0 кДж/моль (2)
Расчеты энтальпий реакций на основе абсолютных энтальпий реагентов(∆H˚298 = H˚прод. - H˚исх.) невозможны вследствие неопределенности последних. Проблема решается допущением, что энтальпия образования простого вещества в его стандартном состоянии равна нулю.
Тогда энтальпии реакций (1) и (2) равны энтальпиям образования СО2 и H2О, соответственно, из простых веществ:
∆H˚298 = ∆H˚обр298
Энтальпия реакции, при которой в стандартных условиях образуется I моль сложного вещества из простых веществ, называется стандартной энтальпией образования данного вещества и обозначается ∆H˚298 (часто один из индексов опускается).
Стандартные энтальпии образования определены для многих сложных веществ.
В таблице 1 приведены данные для некоторых соединений.
Стандартные энтальпии образования соединений при 298,15˚K из простых веществ (∆H˚298, кДж/моль)
Вещество |
∆Н298о обр. кДж/моль |
Вещество |
∆Н298о обр. кДж/моль |
AgBг (к) |
-100.7 |
NaNO3(k) |
-467.0 |
AgС1 (к) |
-127.2 |
NaOH (к) |
-426.6 |
AgF (к) |
-206.0 |
Na2C03(K) |
-1131.5 |
АgN03(к) |
-124,6 |
NaHS04(K) |
-288.0 |
А1С13(к) |
-704.6 |
Na2S04(K) |
-1388.I |
AIF3 (к) |
-1511.4 |
03 (г) |
+142.4 |
А1203(к) |
-1676.8 |
РС13(ж) |
-319.9 |
BaS04(K) |
-1474.2 |
РС15(к) |
-434.6 |
CaC03(K) |
-1207.7 |
PH3 (г) |
+ 22.9 |
СаС12(к) |
-796.3 |
PbCI2(к) |
-360.9 - |
СаС2 (к) |
- 59.9 |
Рb0(к) красный |
-219.4 |
СаО (К) |
-635.5 |
Рb0(к) желтый |
-217.8 |
Са(0Н)2(к) |
-986.8 |
S02(г) |
-297.1 |
HCI (г) |
- 92.4 |
S03 (г) |
-396.I |
СuС12(к) |
-215.7 |
H2S (г) |
- 20.9 |
Сu(0Н)2(к) |
.-444;6 |
H2S04(ж) |
-814.8 |
Сu SО4(к) |
-771.4 |
SnCI2(к) |
-331.2 |
FeCI2(к) |
-342.0 |
Sn С14(ж) |
-529.2 |
FeCI3(к) |
-399.7. |
SnCI4(г) |
-489.4 |
FеС0Н)2(к) |
-562.0 |
С(графит) |
0 |
Fе(0Н)3(к) |
-827.2 |
С (алмаз) |
+ 1.8 |
FeS04 (к) |
-929.5 |
СН4 (г) |
- 74.9 |
FeO (K) |
-265.0 |
C2H2(г) |
+226.2 |
Fe2O3(к) |
-822.7 |
C2H4(г) |
+ 52.5 |
MgCI2(K) |
-641.8 |
C2H6(г) |
+ 84.8 |
MgO (к) |
-602. |
CH30H (ж) |
-239.6 |
Mg(0H)2(K) |
-925.3 |
C2H50H (Ж) |
-277.1 |
MgS04(K) |
-1262.6 |
СН3С0СН3(ж) |
-248.3 |
MnO (K) |
-385.4 |
CCI4 |
-135.4 |
МnO2 (к) |
-521.8 |
CHCI3 (ж) |
-132.7 |
NH3 (г) |
- 45.8 |
CH3C00H (ж) |
484.4 |
NH4CI (к) |
-314.4 |
CH3СOOC2H5 (ж) |
-486.6 |
NH40H (ж) |
-361.5 |
|
|
HN03 (ж) |
-174.2 |
|
|
NO (Г) |
+ 90.3 |
|
|
N02(p) |
+ 33.5 |
|
|
N203(г) |
+ 83.3 |
|
|
N205(к) |
- 42.7 |
|
|
H20 (ж) |
- 286.0 |
|
|
H20 (г) |
-242.0 |
|
|
Н202(ж) |
-187.9 |
|
|
H202(г) |
-136.2 |
|
|
NaBr (к) |
-361.6 |
|
|
NaCI (к) |
-411.4 |
|
|
NaF (к) |
-574.0 |
|
|