

2.2 Кислотно-основный характер водородных соединений
Как было показано в гл.1.2.2 на примере
хлороводорода, важным условием, важным
этапом диссоциации молекулярных
водородных соединений в водных растворах
является их дополнительная поляризация
в ходе взаимодействия с растворителем.
Поэтому склонность тех или иных НnЭ
к диссоциации по связи Н–Э должна
зависеть от поляризуемости (деформируемости)
соответствующих молекул с перспективой
перерастания индукционного взаимодействия
НnЭ – Н2О
в донорно-акцепторное. В свою очередь
деформируемость молекул растет за счет
увеличения числа и размера несвязывающих
электронных облаков. Например, все
молекулы галогеноводородов устроены
сходно: ![]()
.
Число неподеленных электронных пар одинаково, но увеличивается протяженность соответствующих орбиталей, растет их деформируемость. В результате дополнительного поляризующего действия молекул воды дипольный момент Н–Г будет меняться все легче. Этому способствует и уменьшающаяся энергия внутримолекулярной связи. В итоге диссоциация НГ усиливается, растут их кислотные свойства:
HF
![]() Н+ + F–;
Кд = 6.8·10–4
Н+ + F–;
Кд = 6.8·10–4
Кислота средней силы сильные кислоты:
HCl
![]() Н+ + Cl–;
Н+ + Cl–;
HBr
![]() Н+ + Br–;
Н+ + Br–;
HJ
![]() Н+ + J–;
Н+ + J–;
В молекулах водородных соединений элементов одного периода (CH4 – NH3 – H2O – HF) размер несвязывающих электронных облаков несколько уменьшается (в молекуле метана их вообще нет), но, в то же время, увеличивается их число:
![]() ,
,
Как следствие, деформируемость молекул растет, соответственно усиливаются их кислотные свойства:
-
СН4 – в воде практически не растворим, электролитом не является;
-
NH3·H2O
 NН4+ + OH–;
Кд
= 1.76·10–5
– аммиак
в водном растворе является слабым
основанием, причем в составе гидрата
диссоциирует не молекула NH3,
а вода;
NН4+ + OH–;
Кд
= 1.76·10–5
– аммиак
в водном растворе является слабым
основанием, причем в составе гидрата
диссоциирует не молекула NH3,
а вода; -
Н2О
 Н+ + ОН–; КW
= 1·10–14
– вода является чрезвычайно слабым
идеальным амфотерным электролитом;
Н+ + ОН–; КW
= 1·10–14
– вода является чрезвычайно слабым
идеальным амфотерным электролитом; -
HF
 Н+ + F–; Кд
= 6.8·10–4 – фтороводород
в водном растворе является слабой
кислотой, диссоциацией воды можно
пренебречь, т.к. HF
распадается на ионы гораздо лучше.
Н+ + F–; Кд
= 6.8·10–4 – фтороводород
в водном растворе является слабой
кислотой, диссоциацией воды можно
пренебречь, т.к. HF
распадается на ионы гораздо лучше.
2.3 Кислотно-основный характер гидроксидов (но)mЭОn
Формула (НО)mЭОn передает состав самых разнообразных гидроксидов кислотного, основного или амфотерного характера16 вне зависимости от принципов их строения:
|
Ионные полимеры |
Ионно-ковалентные полимеры |
Ковалентные молекулярные соединения |
||
|
n = 0 |
n = 0 |
n = 0–4, m = 1–3 Кислородсодержащие кислоты р– и d– элементов (в высоких степенях окисления): HClO = HOCl), HNO2 = (HO)NO, H2SO4 = (HO)2SO2 HMnO4 = (HO)MnO3 |
||
|
m = 1 гидроксиды щелочных металлов: NaOH, KOH |
m = 2 гидроксиды щелочно-земельных металлов: Ca(OH)2, Ba(OH)2 |
m = 2 гидроксиды некоторых s–, p– и d–элементов: Mg(OH)2, Pb(OH)2, Zn(OH)2 |
m = 3 гидроксиды некоторых р– и d–элементов: Al(OH)3, Cr(OH)3, Fe(OH)3 |
|
|
И другие варианты (в том числе и при больших значениях m) |
И другие варианты (в том числе и при больших значениях m и n) |
Среди кислородсодержащих кислот встречаются и немолекулярные вещества |
||
Нетрудно понять, что кислотно-основные свойства гидроксидов зависят от поляризуемости, от соотношения в энергиях связей Н–ОЭ и НО–Э.
-
Сравнивать, анализировать связи НО–Э на предмет вероятности диссоциации в результате взаимодействия с молекулами растворителя – достаточно сложная и многоплановая задача. В то же время связи НО–Э решающим образом влияют на структуры гидроксидов. Например, чем выше электроотрицательность атома Э, ниже ионность соответствующих соединений. Но, чтобы не иметь проблем с необходимостью обсуждать зависимость электроотрицательности элементов от их степени окисления и других тонких факторов (КЧ, χ атомов-партнеров), удобно в качестве критерия, определяющего характер связи Э–О принять поляризующее действие катиона Э. При слабом п/д(Э) (крупные низкозарядные катионы щелочных элементов – Э+) электронное облако иона О2– (строго говоря – атома кислорода в составе иона ОН–) деформируется слабо. В этом случае электронные облака Э+ и ОН– остаются в значительной степени обособленными, не обобществляются – связь НО–Э+ (Э+–ОН) является высокоионной. С увеличением поляризующего действия Эn+ будет усиливаться искажение электронной оболочки аниона, все в большей степени будет наблюдаться взаимное перекрывание атомных орбиталей Эn+ и атома кислорода в составе иона ОН–, что равносильно уменьшению степени ионности (увеличению степени ковалентности связи НО–Э+ (Э+–ОН). По мере усиления этого процесса структура гидроксидов должна постепенно трансформироваться от ионных полимеров к ионно-ковалентным полимерам и в дальнейшем – к ковалентным молекулярным веществам.
-
Именно так обстоит дело, если сравнивать высшие гидроксиды элементов одного периода:
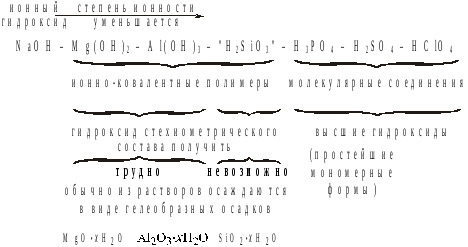
-
В подгруппе, наоборот, за счет роста ионных радиусов п/д(Э) уменьшается и, соответственно, растет степень ионности структур однотипных соединений:
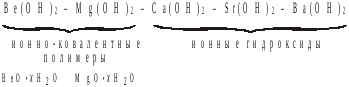
-
Гидроксиды одного и того же элемента так же устроены по-разному в зависимости от его поляризующего действия. Особенно сильно принципы их строения меняются для d–элементов, способных реализовать широкий спектр степеней окисления:

-
Кислотно-основные свойства разнообразных гидроксидов удобно сопоставлять, сравнивая связи Н–ОЭ в их составе. Чем слабей будут эти связи, тем более вероятной окажется их дополнительная поляризация молекулами растворителя (например, воды) с последующей диссоциацией и образованием гидратированных ионов Н+ (Н3О+). В свою очередь, энергия связей Н–ОЭ может отличаться только по причине разного поляризующего воздействия на них соседних катионов Эn+.
-
Сравним, для примера, гидроксиды элементов III-периода. В ряду от NaOH до HClO4 усиливается п/д(Э) на связь Н–ОЭ, соответственно, связь ослабевает – должны постепенно усиливаться кислотные свойства.
– В ионном гидроксиде NaOH связь Н–О наиболее прочна. В то же время кристалл образован за счет электростатических связей (NaOH имеет структуру NaCl). Поэтому диполи воды легче всего ориентируются на заряды ионов и способны вступить с ними в донорно-акцепторное взаимодействие (Na+ – акцептор электронных пар, а гидроксид-ион является потенциальным донором трех электронных пар). Поэтому электролитическому распаду легче всего подвергаются именно межионные связи, это происходит в процессе растворения кристалла. В данном случае межионное притяжение достаточно слабое, NaOH прекрасно растворим в воде, в растворе находится в виде гидратированных ионов. Все это характеризует гидроксид натрия в водном растворе как сильное основание.
– В гидроксидах магния и алюминия межионное притяжение усиливается (уменьшаются ионные радиусы, растут их заряды). Более того, за счет усиления п/д(Э) увеличивается степень перекрывания электронных облаков Ме–О (что приводит к дополнительному упрочению соответствующих связей). В то же время, связи Н–ОЭ в результате усиливающегося п/д(Эn+) ослабевают. В итоге уменьшается основность гидроксидов, что отражается на уменьшении их растворимости, растет вероятность диссоциации по связи Н–ОЭ, т.е. растет вероятность проявления кислотных свойств:

(Под влиянием п/д(Mg2+) ослабевают связи Н–О в молекулах воды,
поэтому небольшая часть аквокомплексов диссоциирует-гидролизуется:
![]()
В итоге:
* Mg(OH)2 весь растворить обычно не удается (ПР ≈ 10–11);
* среда в растворе над осадком не столь щелочная, как это можно было ожидать исходя из растворимости гидроксида;
* в растворе присутствуют, в том числе и ковалентно-связанные группировки, состав которых может быть упрощенно представлен как MgOH+).
![]()
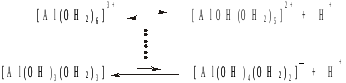
* В растворе над осадком гидроксида алюминия (ПР ≈ 10–32) в целом очень слабо щелочная среда. Но, в то же время, за счет ослабления связей Н–О в составе аквокомплексов в насыщенном растворе Al(OH)3 устанавливается несколько равновесий, участниками которых являются и анионные, и катионные комплексы Al3+. Это позволяет считать гидроксид алюминия амфотерным с преобладанием основных свойств. В сильнокислых средах все равновесия смещаются в сторону катионных аквокомплексов [Al(OH2)6]3+, в сильно щелочных – в сторону анионных гидроксоалюминатов:
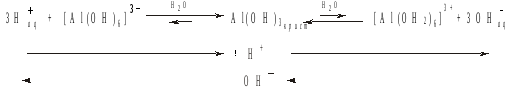
– Гидроксид, формулу которого часто записывают как "H2SiO3", называют кремниевой (кремневой) кислотой. Однако, строго говоря, состав осадка зависит от условий получения, к тому же он постепенно меняется в процессе хранения (обезвоживается). Над осадком может быть обнаружена очень слабокислая среда, где в равновесии с ионами H+ находятся разнообразные, в большинстве случае в той или иной степени полимеризованные силикат-анионы. Катионных аквокомплексов атомы Si+IV не образуют, т.к. их поляризующее действие настолько велико, что молекулы воды в окружении Si+IV не могут сохраниться: партнерами атома кремния в силикатах являются либо атомы кислорода, либо гидроксо-группы, связь Н–О в которых прочней, чем в молекулах Н2О: [SiO2(OH)2]2–, [Si2O7]6–, [Si2O5(OH)2]4–, [Si2O3(OH)4]2– и др. По этим признакам гидроксид кремния SiO2·хН2О следует считать кислотным.
– Кислотами являются и молекулярные гидроксиды Р+V, S+VI и Cl+VII. Но по причине увеличения п/д(Эn+) на связи Н–О в них дополнительно ослабевают: растет сила кислот, все менее вероятно сохранение гидроксо-групп в окружении центрального атома Э+n:
|
Все кислоты молекулярного строения прекрасно растворимы в воде за счет образования межмолекулярных водородных связей. В их разбавленных растворах могут быть обнаружены: |
||
|
в преобладающих количествах: молекулы Н3РО4; далее по мере убывания – ионы: Н+, Н2РО4–, НРО42–, РО43– |
в преобладающих количествах: ионы Н+; далее по мере убывания – ионы: НSО4–, SО42– |
в равных количествах ионы Н+ и ClО4– |
|
Например, молярная концентрация частиц в 0.1М растворах соответствующих электролитов: |
||
|
[H3PO4] = 0.076; [H+] = 0.024; [H2PO4–] ≈ 2.3999999·10–2; [НРО42–] ≈ 5.0·10–8; [РО43–] ≈ 1.1·10–18 |
[H2SO4] = 0; [H+] = 0.11; [HSO4–] = 0.09; [SО42–] = 0.01 |
[HClO4] = 0; [H+] = [ClO4–] = 0.1 |
|
Вывод: ортофосфорная кислота диссоциирует в три стадии; по первой ступени – как кислота средней силы, по второй и третьей стадиям – как слабый электролит.
|
Вывод: серная кислота по первой стадии диссоциирована полностью, да и по второй – достаточно сильно.
|
Вывод: хлорная кислота – сильная одноосновная кислота.
|
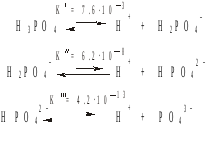

-
В ряду гидроксидов Э(ОН)2 (где Э: элементы подгруппы бериллия) уменьшение п/д(Э2+) приводит к увеличению степени ионности связей Ме–ОН связи, что облегчает электролитический распад по этим связям в процессе растворения: растет растворимость, а вместе с этим усиливаются основные свойства гидроксидов
Ве(ОН)2 – амфотерный гидроксид с преобладанием основных свойств;
Mg(OH)2 – достаточно слабое основание;
Са(ОН)2 – достаточно сильное основание;
Sr(OH)2, Ва(ОН)2 – сильные основания (растворимость и сила растут).
-
В ряду гидроксидов марганца по мере увеличения степени окисления элемента (и одновременного уменьшения радиуса атома Mn+n) усиливается его поляризующее действие на электронные оболочки соседних атомов кислорода (и соответственно – на связи О–Н). Как следствие связи Mn–O становятся все менее ионными (что постепенно затрудняет их электролитический распад), одновременно ослабевают связи О–Н, что, наоборот, увеличивает вероятность диссоциации по кислотному механизму:
|
Mn(OH)2 |
Mn2O3·xH2O |
MnO2·xH2O |
"H2MnO4" |
HMnO4 |
||
|
Основание средней силы |
Амфотерный гидроксид с преобладанием основных свойств |
Амфотерный гидроксид с преобладанием кислотных свойств |
Слабая кислота |
Сильная кислота |
||
|
|
Выполнить кислотно-основные реакции трудно, т.к. сложно предотвратить изменение степеней окисления атома марганца в составе воднорастворимых ионов. |
О кислотных свойствах можно судить по косвенным признакам (по гидролизуемости солей) |
|
|||