

Лекция на 11
.docЛекция на 11-ой неделе (самостоятельно)
Тепловой эффект химической реакции. Энтальпия.
Количество выделившейся или поглощенной теплоты в конкретной химической реакции называется её тепловым эффектом. Тепловой эффект относят либо ко всей реакции [кДж], либо к 1 моль одного из реагентов или продуктов реакций [кДж/моль ], а значение Q записывают со знаком (-) для экзотермической реакции и со знаком (+) для эндотермической реакции. Например:
С(к)+О2(г)=СО2(г) , Q = - 394 кДж или - 394 кДж/моль СО2 .
3О2(г)=2О3(г) , Q = 288 кДж или 144 кДж/моль.
Так как тепловой эффект зависит от агрегатного состояния вещества, его обозначают нижними индексами справа. Например:
H2(г)+Сl2(г)=2HCl (г) Q = - 185 кДж/моль или ΔU = Q = - 185 кДж.
При условии постоянства давление (Р = const) и отсутствует других видов работы
Wм = рΔV, тогда Qр = ΔU+ рΔV или Qр = (U2-U1) +Р(V2-V1) =
(U2+РV2) - (U1+РV1) здесь сумма U+РV – является функцией состояния системы и называется энтальпией H. H = U+РV тогда Qр = H2 - H1 = ΔH :
Qр = ΔU+ рΔV = ΔH
Уравнения химических реакций, записанные с указанием ΔH и агрегатного состояния реагентов, называются термохимическими. ΔH приводят к стандартному состоянию: Р = 1 атм, Т = 298К. В этом случае стандартная энтальпия реакция обозначается символом ΔHº(298К). Например, термохимическое уравнение:
СО(г) +H2O(г) =СО2(г) +H2(г) : ΔHº(298К) = - 41кДж
показывает, что при поддержании давления в 1 атм и температуры 298К будет выделяться 41 кДж теплоты. Для растворенных веществ за стандартное состояние принимают гипотетическое состояние раствора, обладающего свойствами предельно разбавленного раствора с концентрацией вещества “B”, равной 1 моль/л.
Энтальпия образования веществ.
За стандартную энтальпию образования вещества принимают стандартную энтальпию такой реакции, в которой 1 моль этого вещества образуется из простых веществ, каждое из которых находится в термодинамически устойчивом состоянии: символ ΔHº(Т). Например, из двух реакций
С(графит) + O2(г) = СО2(г) : ΔHº(298К) = - 393 кДж
С(к) + O3(г) = СО2(г) + 0,5О2(г) : ΔHº(298К) = - 536 кДж
Только энтальпия первой реакции будет одновременно и стандартной энтальпией образования диоксида углерода.
Стандартная энтальпия образования простого вещества равна нулю. Например: сера S имеет разные аллотропные модификации S8,S6,S2 и др., а углерод – графит, алмаз и карбин. Значения ΔHº(298К) = 0 будет иметь только стабильные модификации: S8 (ромб) и графит.
Закон Гесса и его следствия .
В 1836 г Гесс экспериментально установил: Тепловой эффект реакции, протекающей при Р,Т= const или V,Т= const, зависит только от вида и состояния начальных веществ и продуктов реакции (т.е. от начального и конечного состояния системы), но не зависит от пути реакции и её промежуточных состояний. Закон Гесса показывает, что внутренняя энергия при V, Т = const, равная ΔU=QV и энтальпия при Р,Т = const, Qp = ΔH являются функциями состояния системы. Большое практическое значение для расчета тепловых эффектов реакций имеют следствия из закона Гесса: их 3:
а) изменение энтальпии химической реакции не зависит от числа её промежуточных стадий;
Проиллюстрируем простой схемой. Процесс окисления аммиака до оксида азота может быть осуществлен в одну стадию:
4NH3 (г) + 502 (r) → 4NO(r) + 6Н20(Ж) (1)
∆рΗ°298 = ∆рΗ1 = -1166кДж, или -291,5 кДж/моль NH3 .
Эту же реакцию можно провести в две стадии: окислить аммиак в кислороде с образованием газообразного азота и жидкой воды, а затем азот окислить до монооксида азота:
4NH3 (r) + 302 (r) → 2N2 (г) + 6Н20(Ж)
∆рΗ°298 = ∆рΗ2 = -1531,2кДж, или -382 кДж/моль NH3 ; (2)
2N2(r)+202(r) → 4NO(г),
∆рΗ°298 = ∆рΗ3 = +365,2 кДж или +91,3 кДж/моль NH3 (2.1)
Из сопоставления тепловых эффектов реакций (1) - (2.1) следует, что ∆рΗ1 = ∆рΗ2 + ∆рΗ3. Схематично это представлено рис.1.
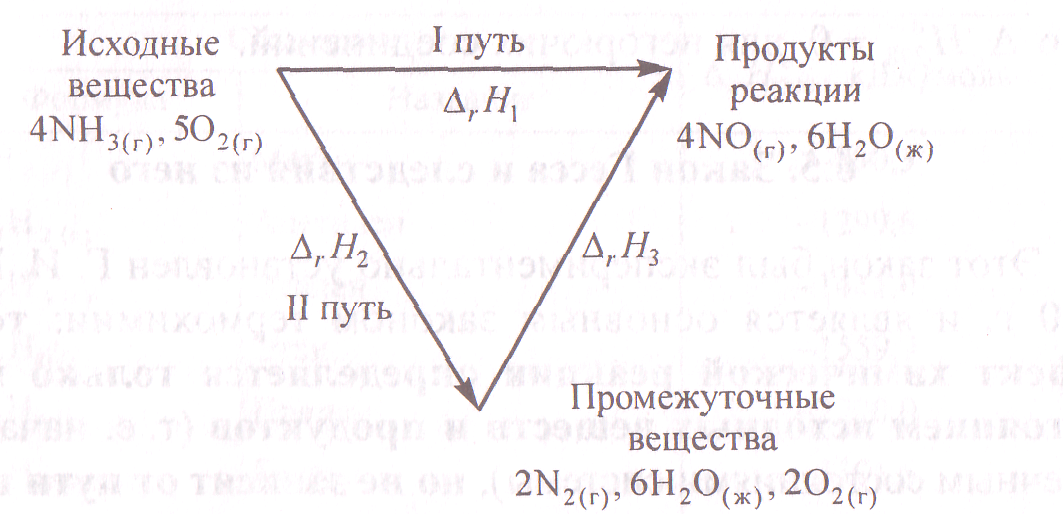
Рис.1. Схема иллюстрации закона Гесса
В простейшем случае мы получаем треугольник, при увеличении числа промежуточных стадий возрастает число сторон многоугольника.
б) энтальпия прямой химической реакции равна взятой с противоположным знаком энтальпии обратной химической реакцией. Например, для реакций: СаСО3(к)=СаО(к)+СО2(г ) ΔH = +178 кДж,
СаО(к)+ СО2(г) =СаСО3(к) ΔH = -178 кДж, значение ΔHº(298К) равно соответственно +178 и -178 кДж.
в) энтальпия реакции равна сумме энтальпий образования продуктов реакции минус сумма энтальпий образования исходных веществ (реагентов):
ΔfHº(298К) = Σ(νjΔf Hº(298К)продуктов) - Σ(νiΔf Hº(298К)реагентов) с учетом стехиометрических коэффициентов. (νj , νi – число молей участвующих в реакции веществ, т.е. стехиометрические коэффициенты).
Например Δf Hº(298К) реакции
PbS(k) + 4H2O2(ж) = PbSО4(к) + 4H2O(ж)
[- 918 + 4*(-286)] - [-94 + 4*(-187)] = - 1220 кДж
стандартные энтальпии образования PbS(k), H2O2(ж), PbSО4, H2O(ж) равны соответственно -94, - 187, -918 и – 286 кДж/моль. Следовательно, данная реакция экзотермическая, выделение теплоты целиком связано с убылью внутренней энергии ΔU, поскольку практически объем системы не меняется.
(Wм = 0, ΔH = ΔU)
Энтропия. Термодинамическое равновесие.
Для оценки принципиальной осуществимости конкретной реакции необходимы специальные количественные критерии. В качестве такого критерия Клаузиусом была предположена функция названная энтропией S (превращение) эта функция равна:
ΔS = S2 - S1 = Q/T (5),
где Q - энергия, которой система в форме теплоты обмениваются с внешней средой при Т - температуре источника теплоты (системы или внешней среды). При наступлении равновесия температуры системы и внешней среды равны. Уравнение (5) получено из анализа работы идеальной машины (цикла Карно).
Энтропия- это свойство, присущее любому веществу, любой системе, так же, как Р, Т, V, U. При термодинамически обратимых процессах в изолированных и адиабатических системах Q = 0 , S2 = S1 и ΔS = 0 то – есть, энтропия их не изменяется, система находится в состоянии термодинамического равновесия (S = Smax , энтропия достигла своего максимума).
Процесс называется термодинамически обратимым, если в любое время химическая реакция может пойти в обратном направлении при бесконечно малом внешнем воздействии и не изменяемой внутренней энергии. В термодинамически необратимом процессе, протекающем в изолированной системе, энтропия возрастает ΔS > 0,
S→ Smax. Для любой химической реакции, протекающей в изолированной системе произвольно и необратимо, энтропия продуктов реакции всегда больше энтропии исходит веществ. Для закрытых систем, находящихся в тепловом равновесии с внешней средой
Т*ΔS = Q (6)
Для необратимых самопроизвольных химических реакций в закрытых системах.
ΔS > Q/ Т.
Экспериментально показано, что энтропия ΔS возрастает сильнее, чем отношение Q/ Т, из-за потерь энергии в форме теплоты, отношение Q/Т представляет собой только минимальное увеличение энергии, соответствующее только термодинамически обратимому процессу.
В общем случае для изолированных систем ∆S > Q, а для закрытых ТΔS > Q.
Второй закон термодинамики.
Второй закон термодинамики является законом возрастания энтропии в необратимых химических реакциях, что доказано опытом работы тепловых машин. Для химически реагирующих систем в связи с участием большого числа частиц вводят понятие вероятности состояния системы. Обычное состояние системы характеризуемое термодинамическими параметрами (Р,Т,V,Cв) называют макросостоянием. Но система с большим числом частиц, у которых непрерывно изменяются энергия, координаты в пространстве, масса и скорость движения, характеризуется понятием микросостояние.
Число микросостояний, с помощью которых осуществляется данное макросостояние системы, называют термодинамической вероятностью (w). Величайшей заслугой Больцмана является получение уравнения:
S= К·lnw (7),
где w- термодинамическая вероятность, постоянная Больцмана – К- коэффициент пропорциональности К = R/Na = 1.38 ∙10^-23 [Дж/молекула∙К]. Из уравнения (7) ясен физический смысл энтропии.
Энтропия-это мера термодинамической вероятности состояния веществ и систем. Уравнение (7) является формой математической формулировки второго закона термодинамики : «любая изолированная система , предоставленная самой себе, изменяется в направлении состояния, обладающего максимальной вероятностью» ΔS= S2- S1=К·lnW2+К·lnW1= К·ln(W1/ W2) : при условии ΔS = 0, W2 = W1, W- термодинамическая вероятность.
Необратимый процесс смешения двух реагирующих веществ, связан с увеличением энтропии системы.
ΔS= S2 - S1 = К·lnW2 + К·lnW1= К·ln W1/ W2 > 0,
но W2 > W1, и тогда ΔS > 0.
Энтропия является мерой хаотичности движения в системе, мерой молекулярного беспорядка. Наиболее беспорядочное статистическое расположение частиц в системе отвечает её наиболее вероятному состоянию. Так, энтропия возрастает в ряду состояний кислорода О > O2 > O3, что связано с увеличением от атомного кислорода до озона числа поступательных, колебательных и вращательных движений частиц вещества.
Энтропия возрастает во всех процессах, сопровождающихся усилении беспорядочного движения частиц вещества при нагревании, сублимации, плавление, диссоциации, дроблении, растворении. Энтропия аморфных веществ больше чем кристаллических.
У твердых веществ энтропия меньше, чем у мягких. Наименьшей энтропией среди простых веществ обладает алмаз – самое твердое вещество.
В совершенно чистом бездефектном кристалле вещества существует абсолютный порядок w = 1, S = К·lnw = 0. Данное макросостояние достигается единственным микросостоянием. Изменение стандартной энтропии химической реакции Δ Sº(298К) определяется, как изменение энтальпии резкостью суммарной энтропии продуктов реакции и суммарной энтропии исходных веществ. (8) ΔS(298К) = Σ[Sº(298К)прод] - Σ[Sº(298К)реаг.]
Третий закон термодинамики. Расчет абсолютных значений стандартных энтропии веществ
В отличие от абсолютных значений внутренней энергии (U) и энтальпии (Н), которые невозможно вычислить, абсолютные значения энтропии (S) поддаются определению и для сложных, и для простых веществ из-за наличия у них начальной точки в шкале отсчета, устанавливаемой третьим законом термодинамики. Третье начало термодинамики иначе называют постулатом Планка: при температуре абсолютного нуля (Т = О К) энтропия идеальных кристаллов любого простого вещества или соединения равна нулю: lim S = 0 , или S0 = 0.
Т->0
Предполагается, что в чистом, не имеющем дефектов кристалле вещества существует абсолютный порядок в расположении частиц, при Т= 0К возможно единственное состояние системы, при котором частицы «застывают» в узлах кристаллической решетки, термодинамическая вероятность равна минимальному значению (W=1), поэтому для одного моля вещества S0 = RlnW = R ln 1 = 0.
Для неидеальных кристаллов, смесей, твердых растворов, стеклообразных структур всегда S0 > 1, иными словами, у них существует так называемая нулевая энтропия, связанная с дефектами кристаллической решетки, возможностью различной ориентации частиц в пространстве и другими причинами.
Так, в узлах молекулярной кристаллической решетки монооксида углерода, молекулы СО могут располагаться двумя способами: СО ОС и СО СО. Если бы обе структуры были равновероятны, то термодинамическая вероятность системы при Т= 0К W = 2, следовательно, S0 = R ln2 = 5,76 Дж/(моль • К). В действительности W < 2, так как вероятность первой структуры несколько выше (но не намного, ибо атомы С и О сходны по электронному окружению), поэтому S0со = 4,7 Дж/(моль • К) .
В литературе имеются и другие формулировки третьего начала термодинамики. Одна из них называется принципом недостижимости абсолютного нуля: при приближении температуры к абсолютному нулю тепловые свойства тел перестают зависеть от температуры, поэтому абсолютный нуль недостижим. В последние годы в США была достигнута температура Т = 900 нК. Другая формулировка распространена в технической термодинамике: даже при устранении всех тепловых потерь нельзя построить тепловую машину, которая охлаждала бы систему до абсолютного нуля, т. е. имела бы коэффициент полезного действия η = 1:
η =(Т1 –Т2)/ Т1 < 1, так как Т2 ≠ 0К .
Стандартное значение энтропии обозначается символом SТ° (Р = 101325 Па) и может быть определено при любом значении температуры, но для удобства сравнения величин SТ° для разных веществ их определяют, как правило, при стандартных термодинамических условиях (р = 101325 Па, Т = 298 К) и обозначают S0298, Дж/(моль • К). Для газообразного вещества при Т = 298 К стандартное значение энтропии S0298 имеет один моль идеального газа при собственном давлении р = 101325 Па = 1 атм, для конденсированного вещества (жидкого или кристаллического) значением энтропии S0298 обладает один моль этого вещества при внешнем давлении р = 1 атм . Для растворенных веществ и ионов в растворах стандартное состояние отвечает моляльной концентрации Сm(i), равной 1 моль/кг Н2О, но при этом предполагается, что раствор обладает свойствами бесконечно разбавленного (идеального) раствора.
Стандартная энтропия любого вещества является положительной величиной (S0298 > 0), изменения энтропии в процессах (∆S) могут быть положительными, отрицательными или равными нулю.
Энергия Гиббса.
Изолированные системы, где осуществимость процесса легко определяется по росту энтропии, редко встречается в практике. В производстве большинство реакции проводят в закрытых системах, где их самопроизвольное течение может идти согласно второму закону термодинамики как с увеличением так и с уменьшением энтропии, лишь бы произведение Т*ΔS алгебраически было больше количества переданной теплоты Q:
Т*ΔS > Q.
У закрытых систем при Р1Т = const, Qp = ΔH, потому ТΔS > ΔH или
ΔH – Т*ΔS < 0
(H2- H1)-Т*(S2- S1) = ( H2 – Т*S2) - ( H1- Т S1) < 0 .
Поскольку энтальпия и энтропия являются функциями состояния системы, разность G = (H - Т S) называют энергией Гиббса. Её изменение равно
G2- G1 = G = ΔH – Т*Δ S<0.
Для закрытых систем Р1Т = 0 и Δ G <0. Для самопроизвольных, необратимых реакций ΔG

рис 2. Изменение энергии Гиббса закрытой системе Р,Т= const, в необратимой (1) и обратимой (2) химической реакции: X-равновесный состав смеси исходных веществ и продуктов реакции. Стандартное значение энергии Гиббса имеет обозначение ΔG (298К) – это изменение энергии Гиббса в реакции образования 1молль вещества из простых веществ, каждое из которых находится в термодинамически устойчивом состоянии:
Δ fG (298К) = Σ[Δ fG (298К)прод]-Σ[Δ fG (298К) реаг].
По значению энтальпии можно безошибочно определить направление только таких реакций, в которых энтальпия практически не меняется. В этом случае ΔG ~ ΔH следовательно экзотермические реакции будут идти самопроизвольно (ΔH < 0), а эндотермические (ΔH > 0) невозможны. При ΔG = 0 или ΔH= Т*ΔS в системе устанавливаются очень подвижное равновесие путем её стремлением к упорядочиванию Δ S < 0 и ΔH < 0 и стремлением к дезинтеграции, разупорядочению ΔS > 0 и ΔH > 0. При малейшем воздействии равновесие в системе будет смещено
Например, в фазовом равновесии “кристалл расплав” понижение температуры вызовет появление новых кристаллов. Зависимость изменения Гиббса от температуры дается соотношением ΔG = ΔH - Т*ΔS, которое приближенно выражается прямой линией. Наклон её зависит от знака и значения ΔS реакции, ΔS > 0 прямая идет вниз, ΔS < 0 прямая идет вверх.
ЛЕКЦИЯ №
Тема: Скорость химических реакций. Химическое равновесие. Скорость химических реакций.
-
Химическое равновесие
-
Энергия активации реакций
-
Константа равновесия реакции.
-
Принцип Ле Шателье
Скорость химических реакций измеряется количеством вещества, вступающего в реакцию или образующегося в результате реакции в единицу времени в единице объема системы (для гомогенной реакции) или на единице площади поверхности раздела фаз (для гетерогенной реакции).
В случае гомогенного процесса, протекающего при постоянном объеме, скорость гомогенной химической реакции измеряется изменением концентрации какого-либо из реагирующих веществ за единицу времени. v = ±∆C/ ∆t , где знак «плюс» относится к изменению концентрации вещества, образующегося в результате реакции (∆С>0), а знак «минус» - к изменению концентрации вещества, вступающего в реакцию (∆С<0).
Зависимость скорости реакции от концентрации определяется законом действия масс: при постоянной температуре скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ.
Так, для реакции типа А+В2→АВ2 закон действия масс выражается следующим образом:
v=k[A][B2].
В этом уравнении [A] и [B2] – концентрации вступающих в реакцию веществ, а коэффициент пропорциональности k – константа скорости реакции, значение которой зависит от природы реагирующих веществ.
А+2В→АВ2 А+В+В→АВ2
В этом случае, в соответствии с законом действия масс, можно записать
v=k[A][B][B], т.е. v=k[A][B]2.
Одновременное столкновение более чем трех частиц крайне маловероятно. Поэтому реакции, в уравнения которых входит большое число частиц (например, 4HCl+O2=2Cl2+2H2O) протекают в несколько стадий, каждая из которых осуществляется в результате столкновения двух (реже трех) частиц. В подобных случаях закон действия масс применим к отдельным стадиям процесса, но не к реакции в целом.
При гетерогенных реакциях концентрации веществ, находящихся в твердой фазе, обычно не изменяются в ходе реакции, поэтому не включаются в уравнение закона действия масс.
Зависимость скорости реакции (или константы скорости реакции) от температуры может быть выражена уравнением:
vt+10/ v t = kt+10/ k t=γ
Здесь vt и кt – скорость и константа скорости реакции при температуре t0C; vt+10 и кt+10 – те же величины при температуре(t+100C); γ – температурный коэффициент скорости реакции. (правило Вант-Гоффа). В общем случае, если температура изменилась на ∆tС, последнее уравнение преобразуется к виду:
vt+∆t / v t =kt+∆t / k t=γ∆t/10
Каждая реакция характеризуется определенным энергетическим барьером; для его преодоления необходима энергия активации – некоторая избыточная энергия (по сравнению со средней энергией молекул при данной температуре), которой должны обладать молекулы, для того чтобы их столкновение было эффективным, т.е. привело бы к образованию нового вещества. С ростом температуры число активных молекул быстро увеличивается, что и приводит к резкому возрастанию скорости реакции.
Зависимость константы скорости реакции k от энергии активации (Еа ,Дж/моль) выражается уравнением Аррениуса:
K=Zpe-Ea/RT
Здесь Z – число столкновений молекул в секунду в единице объема; е – основание натуральных логарифмов (у = 2.718…); R – универсальная газовая постоянная (8.314 дж*моль-1*К-1); Т- температура, К ; Р – так называемый стерический множитель.
Как следует из уравнения Аррениуса, константа скорости реакции тем больше, чем меньше энергия активации.
Скорость хим. реакции возрастает в присутствии катализатора. Действие катализатора объясняется тем, что при его участии возникают нестойкие промежуточные соединения (активированные комплексы), распад которых приводит к образованию продуктов реакции. При этом энергия активации реакции понижается и активными становятся некоторые молекулы, энергия которых была недостаточна для осуществления реакции в отсутствие катализатора. В результате общее число активных молекул возрастает, и скорость реакции увеличивается.
При протекании химической реакции концентрации исходных веществ уменьшаются; в соответствии с законом действия масс это приводит к уменьшению скорости химических реакций. Если реакция обратима, т.е. может протекать как в прямом, так и в обратном направлениях, то с течением времени скорость обратной реакции будет возрастать, т.к. увеличиваются концентрации продуктов реакции. Когда скорости прямой и обратной реакции становятся одинаковыми, наступает состояние химического равновесия и дальнейшего изменения концентрации, участвующих в реакции веществ не происходит.
В случае обратимой химической реакции
А+В↔С+D
Зависимость скоростей прямой (v→) и обратной (v←) реакций от концентраций реагирующих веществ выражается соотношениями:
v→ = k→[A][B]; v← =k ←[A][B].
В состоянии химического равновесия v→ = v←, т.е. k→[A][B]=k←[С].
k→/k←=[C][D]=K.
[A][B]
Здесь К – константа равновесия реакции.
Концентрации, входящие в выражение константы равновесия, называются равновесными концентрациями. Константа равновесия – постоянная при данной температуре величина, выражающая соотношение между равновесными концентрациями продуктов реакции (числитель) и исходных веществ (знаменатель). Чем больше константа равновесия, тем «глубже» протекает реакция, т.е. тем больше выход ее продуктов.
В химической термодинамике доказывается, что для общего случая химической реакции
aA + bB +...→ cC + dD +...
справедливо аналогичное выражение для константы равновесия реакции:
K= [C]a[D]b…
[A]а[B]b…
В выражение константы равновесия гетерогенной реакции, как и в выражение закона действия масс, входят только концентрации веществ, находящихся в жидкой или в газообразной фазе, т.к. концентрации твердых веществ остаются, как правило, постоянными.
Катализатор не влияет на значение константы равновесия, поскольку он одинаково снижает энергию активации прямой и обратной реакций и поэтому одинаково изменяет скорости прямой и обратной реакций. Катализатор лишь ускоряет достижение равновесия, но не влияет на количественный выход продуктов реакции.
При изменении условий протекания реакции (температуры, давления, концентрации какого-либо из участвующих в реакции веществ) скорости прямого и обратного процессов изменяются неодинаково, и химическое равновесие нарушается. В результате преимущественного протекания реакции в одном из возможных направлений устанавливается состояние нового химического равновесия, отличающегося от исходного. Процесс перехода от одного равновесного состояния к новому равновесию называется смещением химического равновесия. Направление этого смещения подчиняется принципу Ле Шателье:
Если на систему, находящуюся в состоянии химического равновесия, оказать какое-либо воздействие, то равновесие сместится в таком направлении, что оказанное воздействие будет ослаблено.
Константа равновесия Кт химической реакции связана со стандартным изменением энергии Гиббса этой реакции ∆G0т уравнением: ∆G0т=-2.3RT lgKт.
При 298К (25С) это уравнение преобразуется к виду: ∆G0298=-5.69 lgK298,
где ∆G0298 выражено в кДж/моль.
Как показывают последние уравнения, отрицательный знак ∆G0 возможен только в том случае, если lgk<0, т.е. K<1. Это значит, что при отрицательных значениях ∆G0 равновесие смещено в направлении прямой реакции и выход продуктов реакции сравнительно велик; при положительном знаке ∆G0 равновесие смещено в сторону обратной реакции и выход продуктов прямой реакции сравнительно мал. В связи с этим следует подчеркнуть, что знак ∆G0 указывает на возможность или невозможность протекания реакции только в стандартных условиях, когда все реагирующие вещества находятся в стандартных состояниях. В общем же случае возможность (или невозможность) протекания реакции определяется знаком ∆G , а не ∆G0