

ximia
.docx1) 1. Основные понятия темы Специфические реакции позволяют обнаруживать ион в отдельной порции анализируемого раствора, не считаясь с присутствием других ионов. При этом последовательность обнаружения ионов может быть произвольной. Дробным анализом называют обнаружение ионов с помощью специфических реакций в отдельных порциях анализируемого раствора, производимое в любой последовательности. Дробный анализ применяют агрохимические и заводские лаборато рии, особенно в тех случаях, когда состав исследуемого материала до статочно хорошо известен и требуется только проверить отсутствие некоторых примесей. Если же используемые реакции не специфичны, а мешающее дейст вие посторонних ионов устранить не удается, то проведение дробного анализа невозможно. В этом случае применяют систематический ход анализа. Сисmемаmическим ходом анализа – называется определенная последовательность выполнения аналитических реакций, при которой каждый ион обнаруживают после того, как будут обнаружены и удалены другие ионы, мешающие его обнаружению. Допустим, что раствор нужно испытать на присутствие катиона Са2+, но в нем одновременно может содержаться и ион Ва2+. Катион Са2+ принято обнаруживать в виде оксалата: CaC12 + (NH4)2С2О4 = СаС2О4 + 2NH4Cl Эта реакция достаточно чувствительна, но не специфична, так как окса лат аммония (NH4)2C204 дает белый кристаллический осадок не только с Са2+, но также с Ва2+ и некоторыми другими ионами. Поэтому преж де чем обнаруживать катион Са2+, необходимо проверить, присутствует ли в растворе мешающий ион Ва2+. Последний можно обнаружить в отдельной порции раствора, действуя хроматом калия, с которым Ва2+ дает характерный желтый осадок: BaCl2 + К2Сг04 = ВаСг04↓ + 2KCl Присутствие иона Са2+ не мешает обнаружению иона Bа2+ этой реакцией, так как хромат кальция СаСгО4 хорошо растворим в воде (выпадает в осадок только из очень концентрированных растворов солей кальция). Дальнейший ход анализа зависит от результата проведенного испы тания. Если окажется, что ион Bа2+ отсутствует, то в другой порции раствора можно обнаруживать катион Са2+, действуя оксалатом аммо ния (NH4)2С2О4. Если же катион Bа2+ присутствует, то прежде чем обнаруживать Са2+, следует полностью удалить из раствора ионы Bа2+. Для этого на весь раствор действуют избытком хромата калия К2Сr04 (или дихромата калия К2Сr207), убеждаются, что ионы Bа2+ полностью осаждены в виде хромата бария BaСrО4 и, отделив осадок, беспрепятственно обнаруживают катионы Са2+. Следовательно, в систематическом ходе анализа применяют не только реакции обнаружения отдельных ионов, но также и реакции отделения их друг от друга. Разделение ионов чаще всего основывается на различной раствори мости аналогичных солей (например, ВаСr04 и СаСrО4). Иногда в этих целях используют и различную летучесть соединений. Так, отделение катиона NH4+ от ионов Na+, К+ и Mg2+ осуществляют выпариванием раствора и прокаливанием сухого остатка. При этом непрочные соли аммония разлагаются, улетучиваются, и соединения Na+, К+ и Mg2+ освобождаются от мешающих примесей этих солей, отделяя один ион от другого, нужно внимательно следить за пол нотой этого разделения, без которой результаты анализа будут оши бочными. Например, при неполном удалении иона NH4+ можно в даль нейшем „переоткрыть” К+ и Nа+, так как с реактивами на эти катионы взаимодействуют и соли аммония. Полноту удаления мешающего иона проверяют в каждом случае специальной пробой. Систематический анализ не следует противопоставлять дробному: эти методы взаимно дополняют друг друга. Каждый из них имеет свою область применения. 2. Макро-, полумикро-, микро- и ультрамикроанализ В зависимости от количества исследуемого вещества, объема раствора и техники выполнения операций аналитические методы качественного анализа подразделяют на макро-, микро- и полумикрометоды. 1.Макрометод - наиболее старый метод химического анализа, при котором для анализа берут сравнительно большие количества вещества и реактивов: 1 г сухого вещества или 20-30 мл реактива, 2.Микрометод - при этом методе для исследования берут примерно в 100 раз меньшие количества вещества, чем в макрометоде: 5-10 мг сухого вещества или 0.2-0.3 мл раствора, При микрометоде используют высокочувствительные реакции – микрокристаллоскопические и капельные, которые проводят на предметном стекле, а о наличии определяемого вещества судят по форме кристаллов, рассматривая их под микроскопом, 3.Полумикрометод - занимает промежуточное положение между макро- и микрометодом. Для анализа берут: 50 мг сухого вещества или 0.1-1.0 мл раствора. Применяют в основном те же реакции, что и при макрометоде, но выполняют их с меньшим количеством реагирующих веществ. Этот метод анализа наиболее удобен, так как требует наименьших затрат, времени, реактивов и оборудования. 3. Техника выполнения важнейших операций в качественном анализе При проведении качественного анализа полумикрометодом приходится выполнять множество операций, требующих определенных навыков. К таким операциям относятся: осаждение ионов, нагревание растворов, центрифугирование, выпаривание, промывание осадка и т.д. 1.Нагревание При нагревании пробирок с большим количеством растворов на спиртовке (или горелке) жидкость может быть выброшена паром. Поэтому лучше всего нагревание вести на слабо кипящей бане. 2.Осаждение Реакции осаждения ионов в форме труднорастворимых соединений применяются в качественном анализе с целью: 1.отделения катионов одних аналитических групп от других; 2.выделения ионов данного элемента из смеси или удаления мешающих ионов; 3.обнаружения отдельных ионов. При проведении реакций осаждения для отделения катионов той или иной аналитической группы следует иметь в виду, что катионов этой группы может совсем не быть в исследуемом растворе, поэтому необходимо реакцию осаждения сначала проделать в отдельной пробе с 2-3 каплями анализируемого раствора. При положительном результате этой пробы следует провести осаждение ионов данной группы из большого объема исследуемого раствора. Техника осаждения следующая: налить в пробирку несколько капель исследуемого раствора и медленно по каплям приливать к нему соответствующий реактив, перемешивая стеклянной палочкой после добавления каждой капли реактива, что ускоряет выпадение осадка. Дать осадку осесть так, чтобы раствор над осадком стал прозрачным. Далее проверить полноту осаждения, для этого прибавить еще 1 каплю реактива и, если она не вызовет помутнения прозрачного раствора над осадком, то осаждение закончено. В противном случае осаждение надо продолжить, добавив к смеси еще несколько капель реактива. Необходимо помнить, что большой избыток реактива - осадителя может вызвать частичное или полное растворение осадка в результате образования растворимых комплексных соединений. Если осаждаемое вещество может переходить в коллоидное состояние, смесь надо подогреть на водяной бане, так как нагревание способствует процессу коагуляции. 3. Центрифугирование В качественном анализе для отделения осадка от раствора производят центрифугирование с помощью электрической центрифуги. При работе с центрифугой следует выполнять следующие правила: 1.Следить за равномерностью загрузки. Для этого следует брать одинакового размера центрифужные пробирки и наполнять их равными объемами жидкости с осадком, но не более, чем на 2/3 высоты пробирки. При работе только с одной пробиркой в противоположный патрон центрифуги нужно помещать такую же пробирку с равным объемом воды. 2.Перед включением центрифуги обязательно нужно закрыть защитный металлический кожух. 3.Включать и выключать центрифугу необходимо посредством плавного, без рывков поворота реостата. 4.Для отделения кристаллических осадков достаточно 2-х минутное цеитрифугирование; для отделения рыхлых, хлопьевидных осадков – 3-4 минутное. Если за это время отделение осадка не произойдет, то жидкость с осадком необходимо нагреть или добавить к ней 1 каплю раствора сильного электролита, не взаимодействующего с осадком, затем вновь отцентрифугировать. В результате коагуляции коллоидов цеитрифугирование пойдет нормально. 4.Перенесение центрифугата Если осадок, полученный при центрифугировании, очень плотный, то надосадочную жидкость (центрифугат) можно осторожно слить с осадка в другую пробирку. Если осадок рыхлый, центрифугат переносят в другую пробирку с помощью глазной пипетки, стараясь не задеть осадок. Если центрифугата над осадком очень мало, то используют капиллярную гашетку. 5.Промывание осадка Промывание осадка проводят в том случае, если осадок должен быть подвержен дальнейшему анализу и необходимо освободиться от примесей, попавших в него из раствора. В качестве промывной жидкости используют дистиллированную воду или разбавленный раствор осадителя. К осадку в центрифужной пробирке нужно прилить около 1 мл. промывной жидкости и тщательно перемешать ее с осадком тонкой стеклянной палочкой. Затем содержимое пробирки отцентрифугировать, жидкость удалить пипеткой. Операцию промывания повторить 2-3 раза, используя свежие порции промывной жидкости. Когда требуется промывать осадок горячей водой, нужно в пробирку с осадком прилить немного дистиллированной воды, перемешать стеклянной палочкой и нагреть на водяной бане в течение 1-2 минут. Затем отцентрифугировать и удалить центрифугат. 6.Растворение осадка Следует помнить, что отделенный от центрифугата осадок нельзя оставлять на длительное время, так как изменяется его структура и состав, в результате уменьшается растворимость. Поэтому свежеосажденные осадки всегда растворяются легче, чем высушенные. Для растворения к осадку нужно прилить по каплям растворитель, постоянно перемешивая его стеклянной палочкой. Не следует спешить с избытком растворителя. А если осадок растворяется медленно, лучше нагреть его на водяной бане. 7. Выпаривание растворов Выпаривание может быть частичным и полным. Частичное выпаривание проводят с целью повышения концентрации раствора. Полное выпаривание растворителя применяется при удалении летучих веществ (например, аммиака). При этом получают сухой осадок, который затем прокаливают. Выпаривание проводят в фарфоровой чашке или тигле на электрической плитке с асбестовой сеткой. Очень малые количества растворов (1-2 капли) выпаривают на стеклянной пластинке или на часовом стекле, пользуясь предварительно нагретой асбестовой сеткой. Выпаривание с выделение вредных газов и паров (НС1, НNО3, NН3 и т.д. ) следует продолжить под тягой. 8.Прокаливание осадка Прокаливание осадка в качественном анализе проводят с целью удаления органических веществ и аммонийных солей. Прокаливание ведут в той же фарфоровой чашке или тигле, откуда предварительно была выпарена жидкость, на электрической плитке. После окончания прокаливания чашку или тигель переносят с помощью тигельных щипцов на асбестовую сетку и дают остыть. 2)
Качественный химический анализ большей частью основан на реакциях, которые сопровождаются каким-либо характерным внешним эффектом. При этом не имеет большого значения количественная сторона химического процесса и стехиометрическое соотношение элементов, входящих в состав образующихся соединений. Существенными являются две характеристики химических реакций: чувствительность и избирательность.
Чувствительность реакций
Чувствительность реакции выражается двумя взаимно связанными величинами: чувствительностью определения (минимальная концентрация, или предельное разбавление) и абсолютной чувствительностью (открываемый минимум):
m = cV, m = eg
где m — открываемый минимум, мкг, с — минимальная концентрация, т. е. та наименьшая концентрация, которую еще удается обнаружить данным методом, мкг/мл, мкг/г, % и т. п.; V—объем пробы, мл; g — масса пробы, г.
Предельное разбавление характеризуется отношением одной весовой части обнаруживаемого иона или вещества к числу весовых частей анализируемого вещества (основы) или указывается как отношение числа весовых частей обнаруживаемого иона или вещества к 106 весовых частей анализируемого вещества (млн-1, международное обозначение, ррт.). Чувствительность выражается также в %
Значения тис могут изменяться в широких пределах в зависимости от условий реакции. Чувствительность реакции может быть повышена изменением концентрации реагентов, рН среды, порядка смешивания реагентов, экстракцией, флотацией, соосаждением, образованием смешанных соединений, а также путем использования различных методов проведения реакций (капельный анализ, пирохимический анализ и др.) и способов наблюдения продуктов реакции (в обычном световом или электронном микроскопе, в потоке ультрафиолетового света и т. д.). Ниже приведены границы чувствительности различных методов качественного анализа (в %):

5)
Активность и ионная сила раствора - Различие в свойствах концентрированных и разбавленных растворов электролитов связано главным образом со значительным электростатическим взаимодействием ионов в концентрированных растворах. В связи с этим вместо понятия концентрация используется понятие активность. Активность i-й ионной частицы а связана с ее концентрацией с, уравнением: a=fici, где fi - коэффициент активности; его значение определяется концентрациями и зарядами всех ионных частиц раствора Активность является мерой концентрации с учетом электростатических межионных взаимодействий. Связь концентраций ионов и зарядов ионов со значением коэффициента активности ионной частицы fi выражается уравнением: -lg(fi)=k√μ; где k - константа, μ - ионная сила При этом многозарядные ионы оказывают более сильное влияние на значение fi, чем однозарядные. Степень этого влияния выражается ионной силой раствора согласно уравнению: μ=½Σ(c1z12+c2z22c3z32+...+ckzk2)=½Σcizi2 где ci молярная концентрация данной ионной частицы, zi - число ее элементарных зарядов
Ионная сила раствора — мера интенсивности электрического поля, создаваемого ионами в растворе. Полусумма произведений из концентрации всех ионов в растворе на квадрат их заряда. Формула впервые была выведена Льюисом:
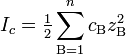 ,
,
где cB — молярные концентрации отдельных ионов (моль/л), zB заряды ионов
Суммирование проводится по всем типам ионов, присутствующих в растворе. Если в растворе присутствуют два или несколько электролитов, то вычисляется общая суммарная ионная сила раствора.
Например, для раствора NaCl с концентрацией 0,001 моль/л, в котором присутствуют два вида однозарядных ионов Na+ и Cl− с концентрациями также равными 0,001 моль/л, ионная сила будет вычисляться следующим образом:
I(NaCl) = 0,5(z²(Na+)•c(Na+) + z²(Cl−)•c(Cl−)) = 0,5(1²•c(NaCl) + (-1)²•c(NaCl)) = c(NaCl)
И ионная сила соответственно будет равна концентрации раствора:
I = 0.5(1²•0,001 моль/л + (-1)²•0,001 моль/л) = 0.5(0,001 моль/л + 0,001 моль/л) = 0,001 моль/л
Это верно для раствора любого сильного электролита, состоящего из однозарядных ионов. Для электролитов, в которых присутствуют многозарядные ионы, ионная сила обычно превышает молярность раствора.
Ионная сила раствора имеет большое значение в теории сильных электролитов Дебая — Хюккеля. Основное уравнение этой теории (предельный закон Дебая — Хюккеля) показывает связь между коэффициентом активностииона ze и ионной силы раствора I в виде:
![]() ,
,
где γ — коэффициент активности, А — постоянная, не зависящая от заряда иона и ионной силы раствора, но зависящая от диэлектрической постоянной растворителя и температуры.
Коэффициенты активности показывают меру отличия свойств реальных растворов от идеальных.
Активности определяют экспериментально, измеряя различные свойства растворов (давление насыщенного пара растворителя над раствором, повышение температуры кипения, понижение температуры замерзания, осмотическое давление и многие другие).
Коэффициенты активности могут быть определены исходя из активности по формуле (7.24). Коэффициенты активности отдельных ионов не могут быть определены экспериментально, но могут быть рассчитаны для разбавленных водных растворов электролитов с помощью уравнения Дебая-Хюккеля:
![]() (7.25)
(7.25)
где γi – коэффициент активности иона; zi –заряд иона; I – ионная сила раствора, определяемая соотношением:
![]() (7.26)
(7.26)
где сi – концентрация и zi –заряд ионов, находящихся в растворе.
Из (7.25; 7.26) следует, что ионная сила раствора, а следовательно и коэффициент активности иона определяются всеми ионами, присутствующими в данном растворе.
Отметим, что уравнение (7.25) справедливо при 0,001<I<0,2. При ионных силах, меньших 0,001 коэффициенты активности мало отличаются от единицы, а при I>0,2 уравнение (7.25) становится слишком грубым приближением.
4)
Электролитическая диссоциация — процесс распада электролита на ионы при его растворении или плавлении.
Классическая теория электролитической диссоциации
Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблуков и В. А. Кистяковский применили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы.
Классическая теория электролитической диссоциации основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциации α, т. е. долей распавшихся молекулэлектролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается законом действующих масс . Например, электролитическая диссоциация бинарного электролита KA выражается уравнением типа:
![]()
Константа
диссоциации ![]() определяется активностями катионов
определяется активностями катионов ![]() , анионов
, анионов ![]() и
недиссоциированных молекул
и
недиссоциированных молекул ![]() следующим
образом:
следующим
образом:
![]()
Значение ![]() зависит
от природы растворённого вещества и
растворителя, а также от температуры и
может быть определено несколькими
экспериментальными методами. Степень
диссоциации (α)
может быть рассчитана при любой
концентрации электролита с помощью
соотношения:
зависит
от природы растворённого вещества и
растворителя, а также от температуры и
может быть определено несколькими
экспериментальными методами. Степень
диссоциации (α)
может быть рассчитана при любой
концентрации электролита с помощью
соотношения:
![]() ,
,
где ![]() —
средний коэффициент активности
электролита.
—
средний коэффициент активности
электролита.
6)
Ио́нное произведе́ние воды́ — произведение концентраций ионов водорода Н+ и ионов гидроксила OH− в воде или в водных растворах, константа автопротолиза воды.
Вывод значения ионного произведения воды
Вода, хотя и является слабым электролитом, в небольшой степени диссоциирует:
![]()
Равновесие этой реакции сильно смещено влево. Константу диссоциации воды можно вычислить по формуле:
![]()
где:
-
[H+] — концентрация ионов гидроксония (протонов);
-
[OH−] — концентрация гидроксид-ионов;
-
[H2O] — концентрация воды (в молекулярной форме) в воде;
Концентрация воды в воде, учитывая её малую степень диссоциации, величина практически постоянная и составляет (1000 г/л)/(18 г/моль) = 55,56 моль/л.
При 25 °C константа диссоциации воды равна 1,8·10−16моль/л. Уравнение (1) можно переписать как:
![]()
Обозначим произведение K·[H2O] = Kв = 1,8·10−16 моль/л·55,56 моль/л = 10−14моль²/л² = [H+]·[OH−] (при 25 °C).
Константа Kв, равная произведению концентраций протонов и гидроксид-ионов, называется ионным произведением воды. Она является постоянной не только для чистой воды, но также и для разбавленных водных растворов веществ. C повышением температуры диссоциация воды увеличивается, следовательно, растёт и Kв, при понижении температуры — наоборот.
[править]Практическое значение ионного произведения воды
Практическое значение ионного произведения воды велико, так как оно позволяет при известной кислотности (щёлочности) любого раствора (то есть при известной концентрации [H+] или [OH−]) найти соответственно концентрации [OH−] или [H+]. Хотя в большинстве случаев для удобства представления пользуются не абсолютными значениями концентраций, а взятыми с обратными знаком их десятичными логарифмами — соответственно, водородным показателем (pH) и гидроксильным показателем (pOH).
Так
как Kв —
константа, при добавлении к раствору
кислоты (ионов H+),
концентрация гидроксид-ионов OH− будет
падать и наоборот. В нейтральной среде
[H+]
= [OH−]
= ![]() моль/л.
При концентрации [H+]
> 10−7 моль/л
(соответственно, концентрации [OH−]
< 10−7 моль/л)
среда будет кислой;
При концентрации [OH−]
> 10−7 моль/л
(соответственно, концентрации [H+]
< 10−7 моль/л) — щелочной.
моль/л.
При концентрации [H+]
> 10−7 моль/л
(соответственно, концентрации [OH−]
< 10−7 моль/л)
среда будет кислой;
При концентрации [OH−]
> 10−7 моль/л
(соответственно, концентрации [H+]
< 10−7 моль/л) — щелочной.
Водоро́дный показа́тель, pH (произносится «пэ аш», английское произношение англ. pH — piː'eɪtʃ «Пи эйч») — мера активности (в очень разбавленных растворах она эквивалентна концентрации) ионов водорода врастворе, и количественно выражающая его кислотность, вычисляется как отрицательный (взятый с обратным знаком) десятичный логарифм активности водородных ионов, выраженной в молях на литр:
![]()
7)
|
Буферные системы, буферные растворы, буферные смеси, системы, поддерживающие определённую концентрацию ионов водорода Н+, то есть определённую кислотность среды. Кислотность буферных растворов почти не изменяется при их разбавлении или при добавлении к ним некоторых количеств кислот или оснований.
Примером буферной системы служит смесь растворов уксусной кислоты CH3COOH и её натриевой соли CH3COONa. Эта соль как сильный электролит диссоциирует практически нацело, т. е. даёт много ионов CH3COO-. При добавлении к буферной системе сильной кислоты, дающей много ионов Н+, эти ионы связываются ионами CH3COO- и образуют слабую (то есть мало диссоциирующую) уксусную кислоту:
Наоборот, при подщелачивании буферной системы, то есть при добавлении сильного основания (например, NaOH), ионы OH- связываются Н+-ионами, имеющимися в буферной системе благодаря диссоциации уксусной кислоты; при этом образуется очень слабый электролит — вода:
По мере расходования Н+-ионов на связывание ионов OH- диссоциируют всё новые и новые молекулы CH3COOH, так что равновесие (1) смещается влево. В результате, как в случае добавления Н+-ионов, так и в случае добавления ОН--ионов, эти ионы связываются и потому кислотность раствора практически не меняется. Кислотность растворов принято выражать так называемым водородным показателем pH (для нейтральных растворов pH=7, для кислых — pH меньше, а для щелочных — больше 7). Приливание к 1 л чистой воды 100 мл 0,01 молярного раствора HCl (0,01 М) изменяет pH от 7 до 3. Приливание того же раствора к 1 л буферной системы CH3COOH + CH3COONa (0,1 М) изменит pH от 4,7 до 4,65, то есть всего на 0,05. В присутствии 100 мл 0,01 М раствора NaOH в чистой воде pH изменится от 7 до 11, а в указанной буферной системе лишь от 4,7 до 4,8. Кроме рассмотренного, имеются многочисленные другие буферные системы (примеры см. в табл.). Кислотность (и, следовательно, pH) буферной системы зависит от природы компонентов, их концентрации, а для некоторых буферных систем и от температуры. Для каждой буферной системы pH остаётся примерно постоянным лишь до определённого предела, зависящего от концентрации компонентов. Примеры буферных систем
Буферные системы широко используются в аналитической практике и в химическом производстве, так как многие химические реакции идут в нужном направлении и с достаточной скоростью лишь в узких пределах pH. Буферные системы имеют важнейшее значение для жизнедеятельности организмов; они определяют постоянство кислотности различных биологических жидкостей (крови, лимфы, межклеточных жидкостей). Основные буферные системы организма животных и человека: бикарбонатная (угольная кислота и её соли), фосфатная (фосфорная кислота и её соли), белки (их буферные свойства определяются наличием основных и кислотных групп). Белки крови (прежде всего гемоглобин, обусловливающий около 75% буферной способности крови) обеспечивают относительную устойчивость pH крови. У человека pH крови равен 7,35—7,47 и сохраняется в этих пределах даже при значительных изменениях питания и др. условий. Чтобы сдвинуть pH крови в щелочную сторону, необходимо добавить к ней в 40—70 раз больше щёлочи, чем к равному объёму чистой воды. Естественные буферные системы в почве играют большую роль в сохранении плодородия полей. В. Л. Василевский.
Начало формы Конец формы |
||||||||||||||||||||||