

Анализ ионного состава воды. Определение неорганических
катионов (NH4+, K+, Na+, Li+, Mg2+, Sr2+, Ba2+, Ca2+).
Особенности методики, практические рекомендации
Здесь будет рассмотрен вариант одновременного определения ряда катионов и анионов с использованием прибора «Капель-103РЕ».
Для определения катионов щелочных и щелочноземельных металлов используют источник высокого напряжения положительной полярности. Это классическая схема (Рис. 7), где детектор находится вблизи катода и ЭОП движется от анода к катоду. В этом случае катионы движутся к катоду в направлении ЭОП, но быстрее него.

Рис. 7. Классическая схема прибора для анализа неорганических
катионов (с положительным высоковольтным блоком).
Для регистрации пиков катионов применяют косвенное детектирование: в состав ведущего электролита вводят поглощающий катион бензимидазола (БИА) в концентрации 0,01 М, обеспечивающей необходимую оптическую плотность исходного раствора. При разделении катионы пробы эквивалентно замещают в растворе катион бензимидазолия, что приводит к снижению оптической плотности в зоне каждого катионного компонента.
При косвенном детектировании электрофореграмма представляет собой базовую линию с отрицательными пиками. Программы обработки данных имеют опцию «Перевернуть» и позволяют представить электрофореграмму в привычном виде с «положительными» пиками.
Бензимидазол в водном растворе является слабым основанием с рКВН+ = 5,8. При рН 5,8 в растворе в равных концентрациях находятся молекулярная и катионная формы бензимидазола, а при рН 4,8 концентрация катионной формы в 10 раз превышает концентрацию молекулярной. Для эквивалентного обмена катионов необходимо поддержание высокой концентрации катионной формы БИА и электролит должен быть слабокислым. При этом резко уменьшается скорость ЭОП, а также возрастает общая концентрация электролита, что приводит к возрастанию тока в капилляре. На практике ведущий электролит готовят на основе винной кислоты, анионы которой обладают малой подвижностью и увеличивают сопротивление электролита, а соотношение кислоты и основания подбирают до достижения необходимого компромисса между временем анализа и величиной тока.
При электрофорезе катионы регистрируются согласно их электрофоретической подвижности. Первыми мигрируют NH4+ и K+, их подвижности одинаковы и обычно они выходят одним пиком. Для разделения NH4+ и K+ в состав ведущего электролита вводят макроцикл 18-краун-6 с гидрофильной внутренней полостью, размер которой соответствует ионному радиусу К+. В результате образуется комплекс включения «гость»–«хозяин». В основе комплексообразования лежат ион-дипольные взаимодействия катиона К+ с атомами кислорода (рис. 8). Благодаря образованию комплекса подвижность К+ снижается, а подвижность других ионов остается без изменений (рис. 9).
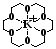
Рис. 8. Комплекс катиона калия с макроциклом 18-краун-6.
Далее с хорошим разрешением идут пики Na+, Li+, Mg2+, Sr2+, Ba2+ и Ca2+.
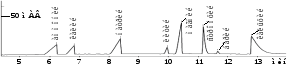
Рис. 9. Электрофореграмма модельного раствора катионов.
«Капель-105», капилляр: внутр. диаметр 75 мкм, Lэфф/Lобщ = 50/60 см.
Ведущий электролит: 10 мМ БИА, 5 мМ винная кислота, 2 мМ 18-краун-6.
Ввод пробы: гидродинамический 30 мбар*10 с. Напряжение: +13 кВ.
Температура: 20°С. Детектирование: 267 нм, косвенное.
Важно знать катионный состав дистиллированной воды, так как от этого будет зависеть возможность и достоверность определения низких концентраций анализируемых катионов (особенно NH4+).
Определение анионов (Cl , SO42, NO2, NO3, F , HPO42)
Для определения анионов в приборе «Капель» необходимо установить источник высокого напряжения отрицательной полярности. Тогда электрод на входном конце капилляра будет катодом, а электрод выходного конца анодом, и анионы будут мигрировать в сторону выходного конца, т. е. к детектору.
На рис. 10 а) показано направление движения ЭОП в противоположную от анионов сторону. Скорость движения анионов заметно превосходит скорость течения жидкости в капилляре, тем не менее, разнонаправленные потоки могут существенно увеличивать времена анализа анионов. Для использования транспортной функции ЭОП, который только переносит зоны разделенных компонентов, не принимая участия в самом процессе разделения, принято обращать направление движения ЭОП (рис. 10б), вводя в состав катионные ПАВ.

Рис. 10. Схемы анализа неорганических анионов.
Ведущий электролит для анализа анионов должен удовлетворять ряду условий.
Во-первых, он должен быть щелочным, так как большинство определяемых анионов существуют только в щелочных средах.
Во-вторых, основой электролита должен быть анион, имеющий сильную полосу поглощения в области 254 нм, так как большинство анионов не обладают собственными полосами поглощения в указанной области, и их определяют только косвенным методом.
В-третьих, ведущий электролит должен содержать добавку для обращения направления электроосмотического потока, так как в противном случае ЭОП, направленный к катоду, резко замедлит или сделает невозможной электромиграцию анионов к детектору.
В-четвертых, катионный компонент ведущего буферного раствора должен быть катионом достаточно сильного основания с малой подвижностью, чтобы обеспечить малую электропроводность раствора.
На практике рабочий буферный раствор состоит из смеси диэтаноламина (основание) и хромовой кислоты с добавкой катионного ПАВ бромида (лучше гидроксида) цетилтриметиламмония (ЦТАБ или ЦТАОН). Избыток диэтаноламина (ДЭА) создает слабо щелочную среду (рН ~9), анион CrO42 обеспечивает необходимое светопоглощение, а катион ЦТА+, сорбируясь на поверхности кварцевого капилляра, перезаряжает поверхность на положительную, чем достигается изменение направления ЭОП.
Бромид цетилтриметиламмония [C16H33(CH3)3N]+Br легко растворим в воде. Как катионное ПАВ, ЦТАБ в малых концентрациях образует истинные растворы, а при более высоких коллоидные. Частицы коллоидного раствора – мицеллы – представляют собой сферические образования, состоящие из 60–100 катионов, обращенных азотным концом наружу, которые несут соответствующий положительный заряд, нейтрализуемый эквивалентным количеством анионов. ККМ для ЦТАБ равна 7 ммоль/л.
Порядок миграции анионов: Cl , NO2, SO42, NO3, F , HPO42. Все пики разрешаются полностью. Выход пика гидрокарбоната, который всегда присутствует в буферном растворе и в растворе пробы, может служить сигналом для окончания анализа.
На электрофореграммах стандартных растворов и растворов проб часто наблюдаются отрицательные пики. Их появление связано с тем, что в растворах проб (стандартов) отсутствуют анионы, которые находятся в растворе ведущего электролита. Первый такой пик может наблюдаться между пиками Cl и NO2 и связан с присутствием в составе ведущего электролита анионов Br (для ЦТАБ в качестве модификатора ЭОП, рис. 11а). Величина этого «бромидного провала» пропорциональна общей концентрации анионов в пробе, и затрудняет автоматическую разметку пика нитрита (исправляется вручную).
Второй отрицательный пик часто наблюдается после пика HPO42. Его появление объясняется тем, что при хранении буферные растворы поглощают все большие и большие количества СО2. В ряде случаев концентрация НСО3 в пробе может оказаться меньше, чем в ведущем электролите, и тогда на электрофореграмме на месте пика НСО3 появляется отрицательный пик. В особых случаях он может быть настолько большим, что будет мешать автоматической разметке пика HPO42.
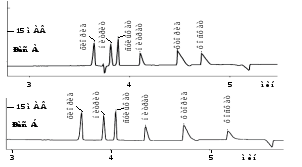
Рис. 11. Электрофоретическое разделение неорганических анионов