

5. Зависимость скорости химической реакции от катализатора.
Катализатор – это вещество, изменяющее скорость химической реакции, количество которого в результате реакции остается неизменным. Изменение скорости химической реакции в присутствии катализаторов называется катализом. Реакции, скорость которых можно изменить при помощи катализаторов, называются каталитическими. Катализаторы, замедляющие скорость химической реакции, называются ингибиторами. Например, тетраэтилсвинец Pb(C2H5)4 – противодействует детонации топлива в двигателях внутреннего сгорания. Вещества, усиливающие действие катализатора называются промоторами. Вещества, подавляющие действие катализатора называются каталитическими ядами; биологические катализаторы называются ферментами.
Механизм катализа может быть различным. По теории промежуточных соединений катализатор (К) с одним из реагирующих веществ (А) образует промежуточное соединение АК ( активированный комплекс ) с более низким значением энергии активации, способное к более эффективному взаимодействию с другим веществом (В).
А
+ В
![]() АВ
АВ
А
+ К ![]() АК
АК
АК
+ В
![]() АВ + К
АВ + К
Таким
образом, катализатор разбивает реакцию
на промежуточные стадии; при этом
образуется ряд неустойчивых промежуточных
соединений – активированных комплексов.
Например, самопроизвольный процесс
разложения пероксида водорода идет
медленно: Н2О2
![]() Н2О
+ 1/2О2
Еа
= 75 кДж/моль .Использование катализатора
MnO2
увеличивает скорость реакции (Еа
= 49 кДж/моль ), за счет образования
активированного комплекса MnO3
:
Н2О
+ 1/2О2
Еа
= 75 кДж/моль .Использование катализатора
MnO2
увеличивает скорость реакции (Еа
= 49 кДж/моль ), за счет образования
активированного комплекса MnO3
:
Н2О2
+ MnO2
![]() H2O
+ MnO3
H2O
+ MnO3
 MnO3
MnO3
![]() MnO2+
1/2O2
H2O2
+ MnO2
MnO2+
1/2O2
H2O2
+ MnO2
![]() H2O
+ MnO2
+ 1/2O2
H2O
+ MnO2
+ 1/2O2
Катализатор снижает энергию активации, разбивая реакцию на ряд промежуточных стадий.
Катализатор может быть специфичен для однотипных реакций, например, V2O5 ускоряет реакции окисления SO2, NH3 и т.д. Он может быть и универсален, т.е. может изменять скорости разных реакций, например,
2SO2
+ O2
![]() 2SO3;
CH2
= CH2
+ H2
2SO3;
CH2
= CH2
+ H2
![]() CH3
– CH3.
Катализатор
может изменять не только скорость
химической реакции, но и ее механизм.
CH3
– CH3.
Катализатор
может изменять не только скорость
химической реакции, но и ее механизм.
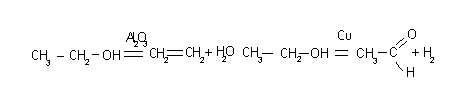
Химическое равновесие.
Когда при химическом взаимодействии хотя бы одно из исходных веществ расходуется полностью, реакцию считают необратимой, протекающей до конца. Например, разложение бертолетовой соли:
KClO3
KCl+ 1,5 O2
.
Многие
химические реакции протекают
обратимо.
Их особенность состоит в том, что они
идут не до конца, в системе всегда
остается (в большем или меньшем количестве)
каждое из исходных веществ. К числу
обратимых реакций относится, например:
СО(Г)
+ Н2О(Г)
= СО2(Г)
+ Н2(Г).
Реакцию, протекающую в правую сторону,
называют прямой,, а в левую – обратной.
Если ![]() прям
=
прям
= ![]() обр,
состояние системы называют химическим
равновесием, его характеризуют
константой равновесия Кр.
обр,
состояние системы называют химическим
равновесием, его характеризуют
константой равновесия Кр.
Кр – это отношение произведения равновесных концентраций продуктов реакции к произведению равновесных концентраций исходных веществ, в степенях их стехиометрических коэффициентов.
Для гомогенных реакций:
aA(г) + bB(г) cC(г) + dD(г), 3Н2+N22NH3
![]() ,
,![]()
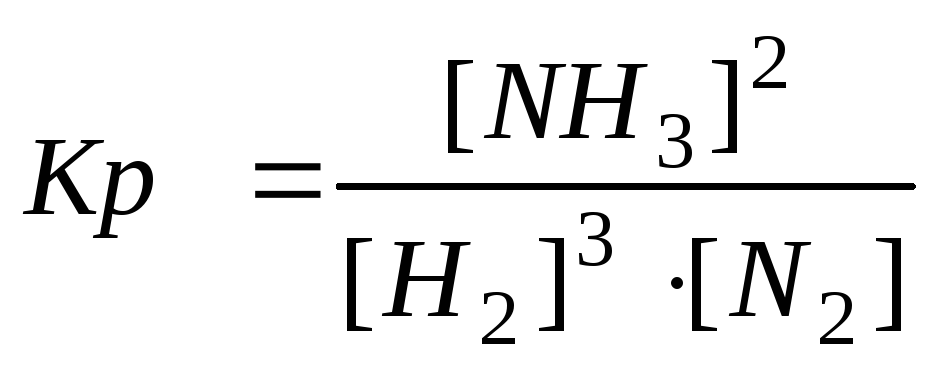
Для гетерогенных реакций:
aA(т) + bB(Г) = cC(Г) + dD(Г) , C(т)+ O2(Г) = СО2(Г)
![]()
![]()
Константа равновесия зависит от природы веществ, температуры. Присутствие катализатора в системе не влияет на значение КР, поскольку он снижает ЕА прямой и обратной реакций на одну и ту же величину и поэтому одинаково изменяет скорости прямой и обратной реакций. Катализатор лишь ускоряет достижение равновесия, он не влияет на количественный выход продуктов реакции. КР не зависит от концентрации реагирующих веществ.
Химическое равновесие является подвижным (динамическим), его можно смещать, изменяя различные факторы (С; Р; T) . Сдвиг химического равновесия определяется принципом Ле-Шателье:
если на систему, находящуюся в равновесии оказывать внешнее воздействие, то система противодействует этому воздействию, стараясь его уменьшить.
Задача.
В
каком направлении произойдет смещение
химического равновесия системы: N2(г)
+ 3H2(г)
![]() 2NH3(г),
ΔΗ‹
0 – (реакция экзотермическая).
2NH3(г),
ΔΗ‹
0 – (реакция экзотермическая).
Реакция идет в прямом Реакция идет в обратном
направлении направлении


если понизить температуру t0С, если повысить t0C
увеличить давлние P уменьшить Р
увеличить
концентрации![]() увеличить
увеличить
![]()
уменьшать
![]() т.е.отводитьNH3
,
т.е.отводитьNH3
,
Задача.
В гомогенной системе СО
+ Сl2
![]() COCl2
, равновесные концентрации
COCl2
, равновесные концентрации
![]() Вычислите Кр и исходные концентрации.
Вычислите Кр и исходные концентрации.
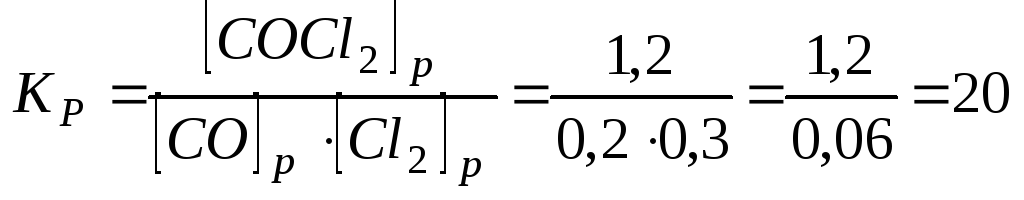 .
.
В результате реакции образовалось 1,2 моля СОСl2, учитывая коэффициенты в уравнении реакции, израсходовалось 1,2 моль/л СО и 1,2 моль/л Сl2, а так как к моменту равновесия осталось 0,2 моль/л СО и 0,3 моль/л Сl2, то
![]()
Рассматривая законы химической термодинамики, было установлено, что возможно протекание тех процессов, у которых ΔG0298<0 (Р, Т = const), а т.к. при химическом равновесии идут обратимые процессы, то какой именно процесс будет преобладать, можно определить по изменению энергии Гиббса. Мы знаем, что равновесие зависит не только от концентрации, но и от температуры и давления (для газообразных систем).
Зависимость Кр. от температуры при постоянном давлении связана с изменением ΔG0298 уравнением:
ΔG0298 = -2,3 RT lg Kр, если R = 8,31·10-3 кДж/моль·К; T = 298 K ΔG0298 = -5,7 lg Kр, кДж/моль.
Если lgKр > 0, т.е. Кр > 1, тогда ΔG0298 < 0 равновесие смещается в сторону прямой реакции.
Если lgKр < 0, т.е. Кр < 1, тогда ΔG0298 > 0 равновесие смещается в сторону обратной реакции.
РАСТВОРЫ ЭЛЕКТРОЛИТОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ.
План
1. Определение понятия «электролиты»
2. Общие представления теории электролитической диссоциации
а) степень диссоциации ()
б) константа диссоциации (Кд)
в) закон Оствальда
3. Электролитическая диссоциация воды
а) ионное произведение воды (Kw)
б) водородный показатель (рН)
в) индикаторы
4. Буферные растворы
5. Гидролиз
Литература
1. Глинка Н.Л. Общая химия: Учебное пособие для вузов. – 24-е изд., исправленное/ Под ред. В.А. Рабиновича. – Л.: Химия, 1985. – с. 225-249.
2. Общая химия. Под ред. Е.М. Соколовской, Г.Д. Вовченко, Л.С. Гузия. М., Изд-во Московского университета, 1980. – с. 239-247, с. 262-268.
3. Лучинский Г.П. Курс химии: Учебник для инженерно-технических (нехимических) вузов. – М.: Высшая школа, 1985. – с. 169-174.
Электролиты – это вещества, которые в растворе или расплаве распадаются на ионы.
Ионы – электрически заряженные частицы, способные к самостоятельному существованию в этих средах.
Электролитическая диссоциация– распад электролита на ионы под действием молекул растворителя.
Для количественной
характеристики электролитической
диссоциации введено понятие степени
диссоциации (![]() ).
).
Степень
диссоциации (![]() )
– это отношение числа (или концентрации
С) молекул распавшихся на ионы, к общему
числу (или к концентрации Сисх)
молекул растворенного электролита.
)
– это отношение числа (или концентрации
С) молекул распавшихся на ионы, к общему
числу (или к концентрации Сисх)
молекул растворенного электролита.
![]()
Электролиты (по заряду иона)
Таблица№12
|
Бинарные или симметричные (распадаются на 2 иона) |
Несимметричные |
|
1,1 валентные: NaCl Na+ + Cl- KBr K+ + Br- |
1,2 валентные: K2SO4 2K+ + SO42- |
|
2,2 валентные: MgSO4 Mg2+ + SO42- ZnSO4 Zn2+ + SO42- |
2,1 валентные: MgCl2 Mg2+ + 2Cl- |
Электролиты (по степени диссоциации)
Таблица№13
|
Сильные
|
Слабые
|
|
Полностью распадаются на ионы.
Обычно это соединения, кристаллическая решетка которых построена из ионов (галогениды щелочных и щелочноземельных металлов NaCl, CaBr2 и т.д.), водные растворы минеральных кислот HCl, HClO4. |
Вещества частично распадаются на ионы.
|
Рассмотрим более подробно разбавленные растворы слабых электролитов. При изучении растворов слабых электролитов (кислот, оснований, солей) было обнаружено некоторое отклонение от законов Вант-Гоффа и Рауля, полученных для растворов неэлектролитов. Для растворов электролитов отмечались более высокие значения осмотического давления, повышение температуры кипения и понижение температуры замерзания по сравнению с растворами неэлектролитов.
Раствор слабого электролита в результате неполной диссоциации представляет собой смесь молекул электролита, ионов электролита, молекул растворителя, между которыми отсутствует взаимодействие.
Таким образом,
отступление от законов Рауля и Вант-Гоффа
обусловлено тем, что количество частиц
в растворе электролита вследствие
неполной диссоциации больше, чем в
растворе неэлектролита. Вант
Гофф для растворов электролитов ввел
поправочный коэффициент
![]() (для неэлектролитов
(для неэлектролитов![]() =1,
для электролитов
=1,
для электролитов![]() >1),
который назвал изотоническим. Уравнения
для электролитов выглядят следующим
образом:
>1),
который назвал изотоническим. Уравнения
для электролитов выглядят следующим
образом:
![]()
![]()
![]()
![]()
Взаимосвязь
изотонического коэффициента (![]() )
со степенью диссоциации (
)
со степенью диссоциации (![]() )
можно найти, используя следующие
рассуждения.
)
можно найти, используя следующие
рассуждения.
Рассмотрим случай,
когда молекула электролита распадается
на
![]() ионов (
ионов (![]() )
со степенью диссоциации
)
со степенью диссоциации![]() .
Тогда количество ионов составляет:
.
Тогда количество ионов составляет:![]() ,
а количество недиссоциированных молекул:
,
а количество недиссоциированных молекул:![]() .
.
Среднее число
частиц, образующихся из одной молекулы
![]() можно записать:
можно записать:
![]() ,
,
![]() ,
,
![]() .
.
Последнее уравнение позволяет вычислить степень диссоциации слабого электролита, если изотонический коэффициент определен экспериментально (например, по изменению температуры замерзания раствора).
Итак, слабый
электролит в растворе диссоциирует на ионы, например, уксусная кислота: СН3СООНСН3СОО-+ Н+
Процесс, обратный диссоциации – рекомбинация ионов в молекулу:
СН3СООН СН3СОО- + Н+
С течением времени устанавливается динамическое равновесие между молекулярной и ионной формами электролита в растворе:
СН3СООН СН3СОО- + Н+
![]() характеризует это
состояние:
характеризует это
состояние:

Константа диссоциации
![]() – это константа равновесия (
– это константа равновесия (![]() ),
отвечающая диссоциации слабого
электролита.
),
отвечающая диссоциации слабого
электролита.
![]() зависит от:
1.Температуры
зависит от:
1.Температуры
2.Природы веществ, растворителя
![]() не зависит от:
не зависит от:
1.Концентрации.
Ступенчатая диссоциация характерна, для слабых многоосновных кислот (например, Н2СО3):
Н2СО3 Н+ + НСО3-
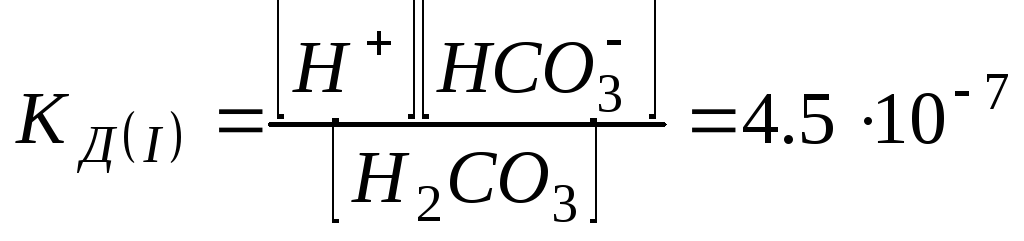 I
ступень
I
ступеньНСО3- Н+ + СО32-
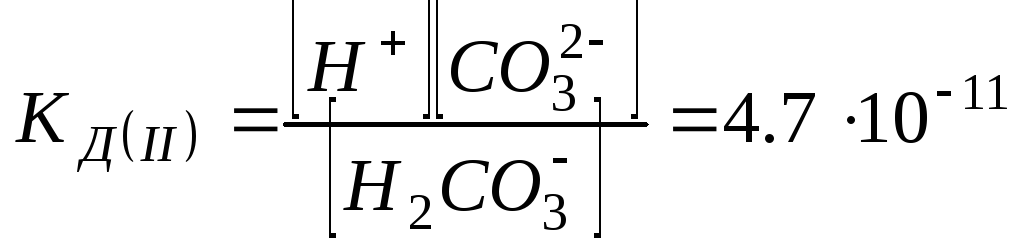 II
ступень
II
ступень
__________________________________________________________
Н2СО3
2Н+
+ СО32-
![]()
Ступенчатая диссоциация для слабых многокислотных оснований:
Fe(OH)3 [Fe(OH)2]+ + OH- KД(I) I ступень
[Fe(OH)2]+ [FeOH]2+ + OH- KД(II) II ступень [FeOH]2+ Fe3+ + OH- KД(III) III ступень
______________________________________________
Fe(OH)3 Fe3+ +3OH- KД = КД(I) КД(II) КД(II
КД(I) > КД(II) > КД(III) (распад происходит в меньшей степени по последующим ступеням).
Взаимосвязь
![]() с
с![]() (закон разведения Оствальда).
(закон разведения Оствальда).
Сисх С С
Рассмотрим пример: СН3СООН СН3СОО- + Н+
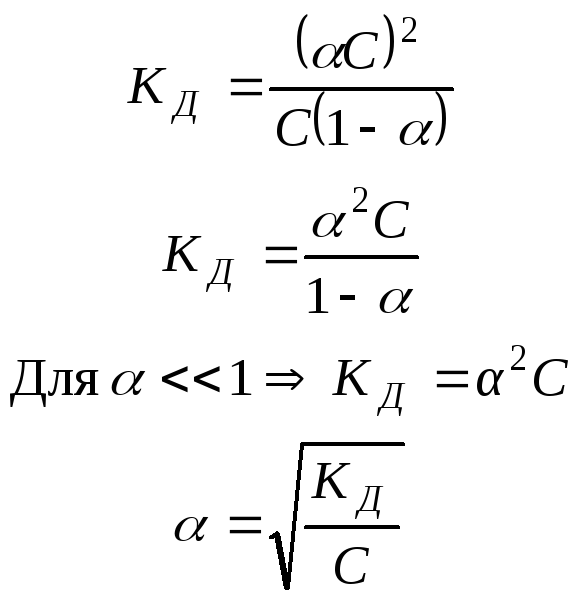
Сисх – исходная концентрация уксусной кислоты СН3СООН
С – концентрация ионов
Сравн – равновесная концентрация уксусной кислоты
Сравн = Сисх – С
Вывод:
1.при разведении раствора, т.е. уменьшении С, увеличивается (приближается к единице)
2.при увеличении С, – уменьшается (вероятность взаимной встречи ионов в растворе и их воссоединения в недиссоциированные молекулы увеличивается)
Электролитическая диссоциация воды. Водородный показатель.
Важной особенностью жидкой воды является ее способность к самопроизвольной диссоциации по реакции:
Н2О(ж) Н+(водн) + ОН-(водн)
Этот процесс называется еще самоионизацией или автопротолизом. Образовавшиеся протоны Н+ и анионы ОН- окружены определенным числом полярных молекул воды, т.е. гидратированы: Н+nH2O; OH-mH2O. Первичная гидратация может быть представлена рядом аквакомплексов: Н3О+; Н5О2+; Н7О3+; Н9О4+, среди которых преобладают ионы Н9О4+ (Н+4H2O). Время жизни всех этих ионов в воде очень мало, т.к. протоны постоянно мигрируют от одних молекул
воды к другим. Обычно в уравнениях для простоты используют только катион состава Н3О+ (Н+H2O), называемый ионом гидроксония.
Процесс диссоциации воды с учетом гидратации протона и образования иона гидроксония может быть записан: 2Н2О Н3О+ + ОН-
Вода – слабый электролит, степень диссоциации которого
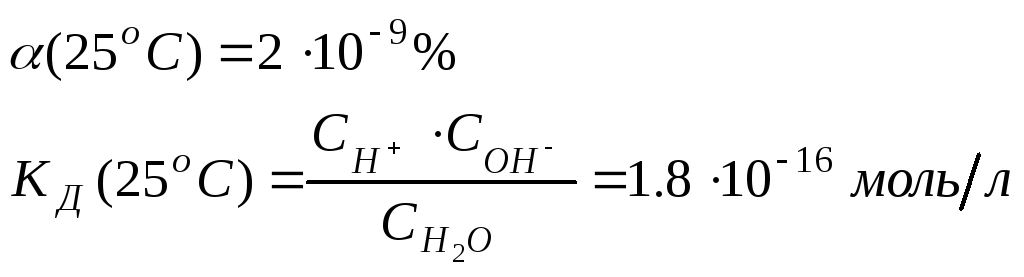
Поскольку
![]()
Сравн(Н2О)
Сисх(Н2О)
или [Н2О]равн
≈ [Н2О]исх
Сравн(Н2О)
Сисх(Н2О)
или [Н2О]равн
≈ [Н2О]исх
![]() –количество молей
содержащееся в одном литре воды. Сисх(Н2О)
в разбавленном растворе остается
постоянной. Это обстоятельство позволяет
включить Сравн(Н2О)
в константу равновесия.
–количество молей
содержащееся в одном литре воды. Сисх(Н2О)
в разбавленном растворе остается
постоянной. Это обстоятельство позволяет
включить Сравн(Н2О)
в константу равновесия.

Таким образом,
произведение двух постоянных величин
![]() дает новую постоянную, которую называютионным
произведением воды
дает новую постоянную, которую называютионным
произведением воды
![]() .
При температуре 298 К
.
При температуре 298 К![]() .
.
- Постоянство ионного произведения воды означает, что в любом водном растворе: кислотном, нейтральном или щелочном – всегда имеются оба вида ионов (Н+ и ОН-)
- В чистой воде концентрации водородных и гидроксильных ионов равны и при нормальных условиях составляют:
[H+] = [OH-] = Kw1/2 = 10-7 моль/л.
- При добавлении кислот концентрация [Н+] увеличивается, т.е. равновесие смещается влево, а концентрация [ОН-] уменьшается, однако Кw остается равной 10-14.
В кислой среде [H+] > 10-7 моль/л, а [OH-] < 10-7 моль/л
В щелочной среде [H+] < 10-7 моль/л, а [OH-] > 10-7 моль/л
На практике для удобства используют водородный показатель (рН) и гидроксильный показатель (рОН) среды.
Это есть взятый с обратным знаком десятичный логарифм концентраций (активностей) соответственно ионов водорода или гидроксильных ионов в растворе: рН = - lg[H+], рОН = - lg[OH-]
В водных растворах рН + рОН = 14.
Таблица№14
|
Растворы | ||
|
Нейтральные |
Кислые |
Щелочные |
|
рН = 7 |
рН < 7 |
рН > 7 |
Kw зависит от температуры (т.к. диссоциация воды – эндотермический процесс)
Kw (25 oC) = 10-14 рН = 7
Kw (50 oC) = 5,4710-14 рН = 6,63
Измерение рН используется чрезвычайно широко. В биологии и медицине величина рН биологических жидкостей служит для определения патологий. Например, в норме рН сыворотки крови состовляет 7,40,05; слюны – 6,35..6,85; желудочного сока – 0,9..1,1; слез – 7,40,1. В сельском хозяйстве рН характеризует кислотность почв, экологическое состояние природных вод и т.д.
Кислотно-основными индикаторами называются химические соединения, изменяющие свою окраску в зависимости от рН среды, в которой они находятся. Вы, наверное, обращали внимание на то, как меняется цвет чая, если в него положить лимон – это пример действия кислотно-основного индикатора.
Индикаторы, как правило, представляют собой слабые органические кислоты или основания и могут существовать в растворах в виде двух таутомерных форм:
HInd H+ + Ind-, где HInd – кислотная форма (это форма, которая преобладает в кислых растворах); Ind – основная форма (преобладает в щелочных растворах).
Поведение индикатора подобно поведению слабого электролита в присутствии более сильного с одноименным ионом. Чем больше [H+] следовательно равновесие смещается в сторону существования кислотной формы HInd и наоборот (принцип Ле-Шателье).
Опыт показывает наглядно возможность использования некоторых индикаторов:
Таблица№15
|
Индикатор |
рН < 7 |
рН = 7 |
рН > 7 |
|
Метилоранж |
красный |
оранжевый |
желтый |
|
Фенолфталеин |
бесцветный |
бесцветный |
малиновый |
|
Лакмус |
красный |
Фиолетовый |
синий |
Специальные приборы – рН-метры позволяют измерять рН с точностью до 0,01 в интервале от 0 до 14. Определение основано на измерении ЭДС гальванического элемента, один из электродов которого является, например, стеклянным.
Наиболее точно концентрацию водородных ионов можно определить методом кислотно-основного титрования. Титрование – это процесс постепенного добавления небольшими порциями раствора известной концентрации (титранта) к титрируемому раствору, концентрацию которого хотим определить.
Буферные растворы – это системы, рН которых относительно мало изменяется при разбавлении или добавлении к ним небольших количеств кислот или щелочей. Чаще всего они представляют собой растворы, содержащие:
а)Слабую кислоту и ее соль(СН3СООН + СН3СООNa) – ацетатный буфер
в)Слабое основание и его соль(NH4OH + NH4Cl) – аммиачно-аммонийный буфер
с)Две кислые соли с разными Kд (Na2HPO4 + NaH2PO4) – фосфатный буфер
Регулирующий механизм буферных растворов рассмотрим на примере ацетатного буферного раствора.
CH3COOH
CH3COO-
+ H+,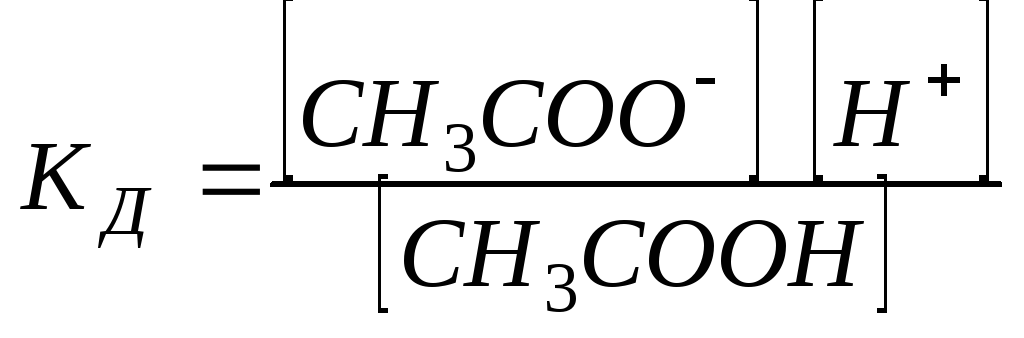
CH3COONa CH3COO- + Na+
1)если добавить небольшое количество щелочи к буферной смеси:
CH3COOH + NaOH CH3COONa + H2O,
NaOH нейтрализуется уксусной кислотой с образованием более слабого электролита H2O. Избыток натрия ацетата смещает равновесие в сторону образовавшейся кислоты.
2)если добавить небольшое количество кислоты:
CH3COONa + HCl CH3COOH + NaCl
Катионы водорода Н+ связывают ионы CH3COO-
Найдем концентрацию ионов водорода в буферном ацетатном растворе:
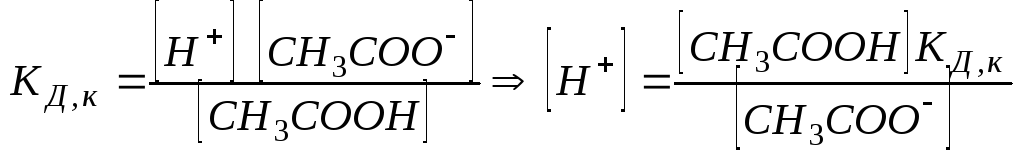
Равновесная
концентрация уксусной кислоты рана
Cисх,к
(т.к. слабый электролит), а [СH3COO--]
= Cсоли
(т.к. соль является сильным электролитом),
то
![]() .
Уравнение Гендерсона-Хассельбаха:
.
Уравнение Гендерсона-Хассельбаха:
![]()
Таким образом, рН буферных систем определяется соотношением концентраций соли и кислоты. При разбавлении это соотношение не меняется и рН буфера не меняется при разбавлении, это отличает буферные системы от раствора чистого электролита, для которого справедлив закон разведения Оствальда.
Существует две характеристики буферных систем:
1.Буферная сила. Абсолютная величина буферной силы зависит от
общей концентрации компонентов буферной системы, т.е. чем больше концентрация буферной системы, тем больше требуется щелочи (кислоты) для одного и того же изменения рН.
2.Буферная
емкость (В).
Буферная емкость – это предел, в котором
проявляется буферное действие. Буферная
смесь поддерживает рН постоянным только
при условии, что количество прибавляемых
к раствору сильной кислоты или основания
не превышает определенной предельной
величины – В. Буферная
емкость определяется
числом
г/экв сильной кислоты (основания), которое
необходимо прибавить к одному литру
буферной смеси, чтобы изменить значение
рН на единицу, т.е.
![]() .
Вывод: Свойства буферных систем:
.
Вывод: Свойства буферных систем:
1.[H+] мало зависит от разбавления.
2.Прибавление сильных кислот (оснований) мало изменяет [H+] в пределах буферной емкости В.
3.Буферная емкость зависит от буферной силы (от концентрации компонентов).
4.Максимальное действие проявляет буфер в случае, когда кислота и соль присутствуют в растворе в эквивалентных количествах:
Ссоли = Ск-ты; [H+] = Кд,к; рН = рКд,к (рН определяется значением Кд).
Гидролиз – это химическое взаимодействие воды с солями. Гидролиз солей сводиться к процессу передачи протонов. В результате его протекания появляется некоторый избыток водородных или гидроксильных ионов, сообщающих раствору кислотные или щелочные свойства. Таким образом, гидролиз обратен процессу нейтрализации.
Гидролиз солей включает 2 стадии:
а)Электролитическая диссоциация соли с образованием гидратированных ионов: KCl K+ + Cl- K+ + xH2O K+ xH2O (связь донорно-акцепторная, донор – атом О, имеющий 2 неподеленные электронные пары,
акцептор – катионы с вакантными орбиталями)
Cl- + yH2O Cl- yH2O (водородная связь)
в) Гидролиз по аниону. Cl- + HOH HCl + OH-
с) Гидролиз по катиону. K+ + HOH KOH +
Гидролизу подвергаются все соли, образованные с участием слабых
электролитов:
1.Соль, образованная анионом слабых кислот и катионом сильных оснований [CH3COONa, NaClO, KNO2, Na2CO3, Na3PO4]
CH3COONa + HOH CH3COOH + NaOH
CH3COO- + НОН CH3COOН + OH-, рН > 7
Анионы слабых кислот выполняют функцию оснований по отношению к воде – донору протонов, что приводит к увеличению концентрации ОН-, т.е. подщелачиванию среды.
Глубина протекания гидролиза определяется: степенью гидролиза г:
![]() ,
,
![]() –концентрация
соли, подвергшейся гидролизу
–концентрация
соли, подвергшейся гидролизу
![]() –концентрация
исходной соли
–концентрация
исходной соли
г невелика, например, для 0,1 моля раствора CH3COONa при 298 К она равна 10-4.
При гидролизе в системе устанавливается равновесие, характеризующееся Кр
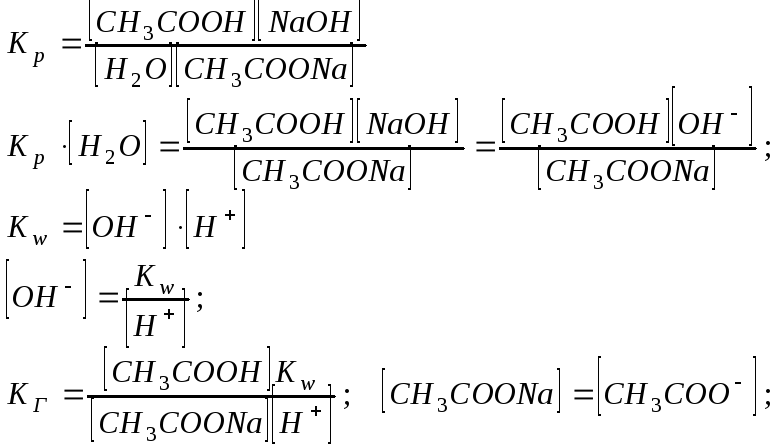
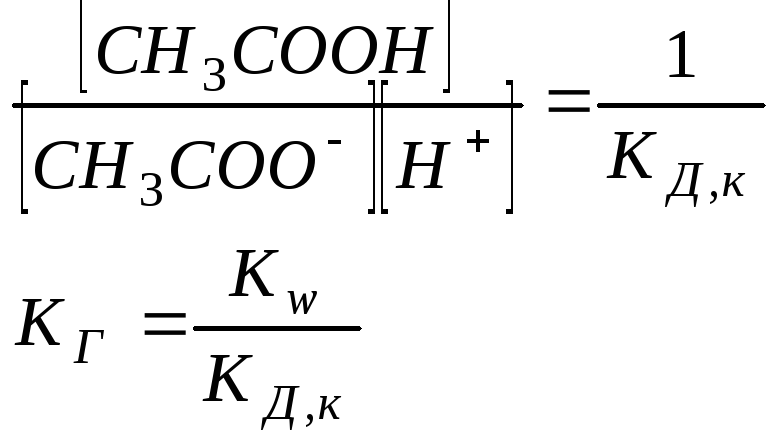
Следовательно, чем меньше константа диссоциации, тем больше константа гидролиза. Степень гидролиза с константой гидролиза связана уравнением:
![]() .
.
С увеличением разбавления, т.е. уменьшением С0, степень гидролиза увеличивается.
2.Соль, образованная катионом слабых оснований и анионом сильных кислот [NH4Cl, AgNO3, ZnCl2, Fe2(SO4)3]
NH4Cl + HOH ↔ NH4OH +
NH4+ + HOH ↔ NH4OH + H+, pH < 7
Протолитическое равновесие смещено влево, катион слабого основания NH4+ выполняет функцию кислоты по отношению к воде, что приводит к подкислению среды. Константа гидролиза определяется по уравнению:
![]()
Равновесная концентрация ионов водорода может быть вычислена: [Н+]равн = г С0 (исходная концентрация соли), где
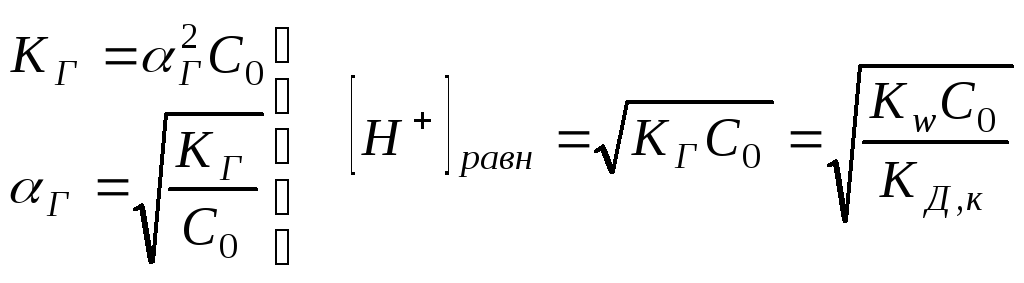 ;
;
![]()
Кислотность среды зависит от исходной концентрации солей подобного вида.
3.Соль, образованная анионом слабых кислот и катионом слабых оснований. Гидролизуется и по катиону и по аниону [NH4CN, CH3COONH4, (NH4)2S, (NH4)2CO3, (NH4)3PO4, ZnS, AlPO4, Al(CH3COO)3, C4(HCOO)2]
NH4CN + HOH NH4OH + HCN
Для определения рН среды раствора сравнивают КД,к и КД,осн
КД,к > КД,осн среда слабо кислая
КД,к < КД,осн среда слабо щелочная
КД,к = КД,осн среда нейтральная
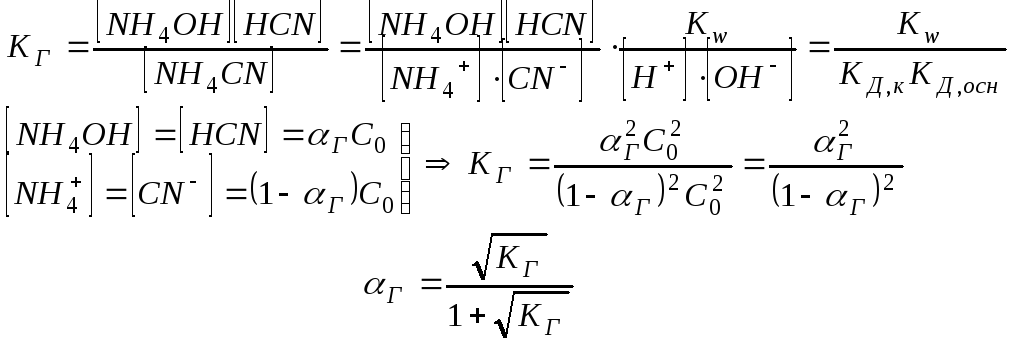
Следовательно, степень гидролиза этого вида солей не зависит от их концентрации в растворе.
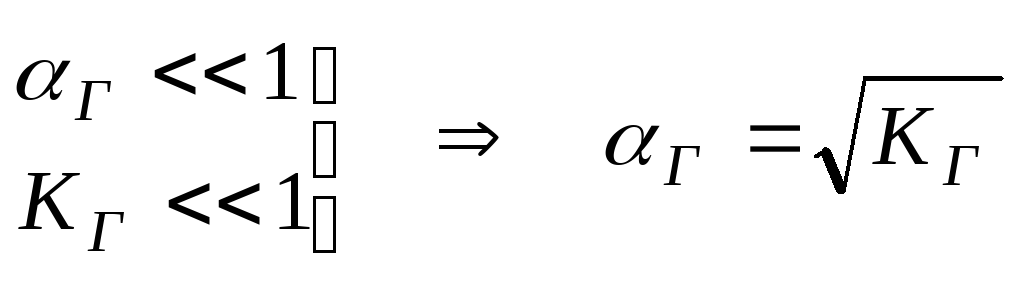
т.к. [H+]
и [ОН-]
определяются КД,к
и КД,осн,
то
![]()
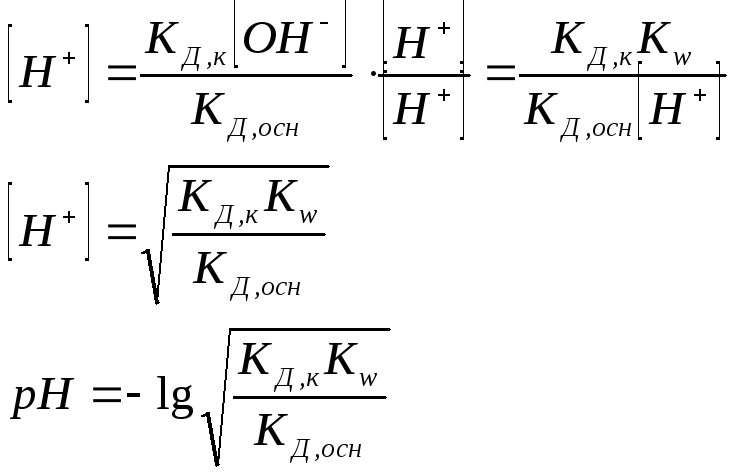
рН раствора также не зависит от концентраций соли в растворе.
Соли, образованные многозарядным анионом и однозарядным катионом (сульфиды, карбонаты, фосфаты аммония) практически полностью гидролизуются по первой ступени, т.е. находятся в растворе в виде смеси слабого основания NH4OH и его соли NH4HS, т.е. в виде аммонийного буфера.
Для солей, образованных многозарядным катионом и однозарядным анионом (ацетаты, формиаты Al, Mg, Fe, Cu) гидролиз усиливается при нагревании и приводит к образованию основных солей.
Гидролиз нитратов, гипохлоритов, гипобромитов Al, Mg, Fe, Cu протекает полностью и необратимо, т.е. соли не выделены из растворов.
Соли: ZnS, AlPO4, FeCO3 и др. в воде малорастворимы, тем не менее часть их ионов принимает участие в процессе гидролиза, это приводит к некоторому возрастанию их растворимости.
Сульфиды хрома и алюминия гидролизуются полностью и необратимо с образованием соответствующих гидроксидов.
4.Соли, образованные анионом сильных кислот и сильных оснований гидролизу не подвергаются [KCl, Na2SO4, Ba(NO3)2].
Чаще всего гидролиз ‑ вредное явление, вызывающее различные осложнения. Так при синтезе неорганических веществ из водных растворов в получаемом веществе появляются примеси – продукты его гидролиза. Некоторые соединения из-за необратимо протекающего гидролиза вообще не удается синтезировать.
-если гидролиз протекает по аниону, то в раствор добавляют избыток щелочи
-если гидролиз протекает по катиону, то в раствор добавляют избыток кислоты
Итак, первая качественная теория растворов электролитов была высказана Аррениусом (1883 – 1887 г.). По этой теории:
1.Молекулы электролита диссоциируют на противоположные ионы
2.Между процессами диссоциации и рекомбинации устанавливается динамическое равновесие, которое характеризуется КД. Это равновесие подчиняется закону действия масс. Долю распавшихся молекул характеризует степень диссоциации . КД и связывает закон Оствальда.
3.Раствор электролита (по Аррениусу) – это смесь молекул электролита, его ионов и молекул растворителя, между которыми отсутствует взаимодействие.
Вывод: теория Аррениуса позволила объяснить многие свойства растворов слабых электролитов при небольшой концентрации.
Однако, теория Аррениуса носила только физический характер, т.е. не рассматривала вопросы:
По какой причине вещества в растворах распадаются на ионы?
Что происходит с ионами в растворах?
Дальнейшее развитие теория Аррениуса получила в работах Оствальда, Писаржевского, Каблукова, Нернста и т.д. Например, на важное значение гидратации впервые указал Каблуков (1891), положив начало развитию теории электролитов в направлении, которое указывал Менделеев (т.е. ему впервые удалось объединить сольватную теорию Менделеева с физической теорией Аррениуса). Сольватация – это процесс взаимодействия электролита
молекулами растворителя с образованием комплексных соединений сольватов. Если растворителем является вода, следовательно, процесс взаимодействия электролита с молекулами воды называется гидратацией, а аквакомплексы – кристаллогидратами.
Рассмотрим пример диссоциации электролитов, находящихся в кристаллическом состоянии. Этот процесс возможно представить в две стадии:
1.разрушение кристаллической решетки вещества Н0кр > 0, процесс образования молекул (эндотермический)
2.образование сольватированных молекул, Н0сольв < 0, процесс экзотермический
Результирующая теплота растворения равна сумме теплот двух стадий Н0раств = Н0кр + Н0сольв и может быть как отрицательной, так и положительной. Например, энергия кристаллической решетки KCl = 170 ккал/моль.
Теплота гидратации ионов К+ = 81 ккал/моль, Cl- = 84 ккал/моль, а результирующая энергия равна 165 ккал/моль.
Теплота гидратации частично покрывает энергию необходимую для выделения ионов из кристалла. Оставшиеся 170 - 165 = 5 ккал/моль могут быть покрыты за счет энергии теплового движения, и растворение сопровождается поглощением теплоты из окружающей среды. Гидраты или сольваты облегчают эндотермический процесс диссоциации, затрудняя рекомбинацию.
А вот ситуация, когда присутствует только одна из двух названных стадий:
1.растворение газов – нет первой стадии разрушения кристаллической решетки, остается экзотермическая сольватация, следовательно растворение газов, как правило, экзотермично.
2.при растворении кристаллогидратов отсутствует стадия сольватации, остается лишь эндотермическое разрушение кристаллической решетки. Например, раствор кристаллогидрата: CuSO4 5H2O (т) CuSO4 5H2O (р)
Нраств = Нкр = + 11,7 кДж/моль
Раствор безводной соли: CuSO4 (т) CuSO4 (р) CuSO4 5H2O (р)
Нраств=Нсольв+Нкр= - 78,2 + 11,7 = - 66,5 кДж/моль
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
План
Значение комплексных соединений (КС).
Координационная теория строения КС А.Вернера. Природа связи в КС.
Классификация КС.
Номенклатура КС.
Пространственное строение КС. Магнитные и оптические свойства КС.
5. Устойчивость КС.
6. Получение и разрушение КС.
Литература.
1. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
2. Курс общей химии под ред. Коровина Н.В. и др. Гл.X, §X.l-X.5.
Комплексные или координационные соединения (КС) составляют наиболее разнообразный класс неорганических веществ, имеющий большое значение в природе и технике. По своему составу КС являются более сложными, чем оксиды, гидроксиды, кислоты и соли. Реакции комплексообразования лежат в основе получения редких и драгоценных металлов, чистых и сверхчистых веществ, аналитических определений, гальванотехнических процессов, получения лекарств, антидетонаторов, антиоксидантов и т.д.
Неоценима роль КС в биологических процессах. Такие жизненно важные природные соединения, как гемоглобин, хлорофилл, инсулин, некоторые витамины относятся также к координационным соединениям.
Строение и механизм образования химических связей в КС наиболее удачно обьясняет координационная теория, предложенная в 1893 г. А.Вернером. Причиной комплексообразования может быть как электростатическое, так и донорно-акцепторное взаимодействие. Рассмотрим механизм образования молекулы [Ag(NH3)2]Cl: AgCl + 2NH4OH [Ag (NH3) 2] Cl + 2H2O
В
молекуле [Ag(NH3)2]Cl
ион серебра имеет следующее строение:
Ag0
(4d105s1)![]() Ag+(4d105s0),
ион хлора -соответственно:
Ag+(4d105s0),
ион хлора -соответственно:
Cl0
(3s23p5)
![]() Cl-
(3s23p6).
Cl-
(3s23p6).
Взаимодействие
катиона Ag![]() c
анионом Cl¯
носит электростатический характер:
Ag+
+
Cl-
c
анионом Cl¯
носит электростатический характер:
Ag+
+
Cl-
![]() AgCl
ионная
связь . При образовании комплексного
иона [Ag(NH3)2]
AgCl
ионная
связь . При образовании комплексного
иона [Ag(NH3)2]![]() катион Ag
катион Ag![]() выступает в качестве акцептора готовой
электронной пары, предоставляя, свободные
орбитали (5s
и 5p)
для координации; молекулы аммиака
выступают в качестве донора электронной
пары. Химическая связь такого вида
называется донорно-акцепторной
или координационной. По своей природе
- это полярная ковалентная связь,
отличие состоит лишь в способе её
образования. Координационные соединения
состоят из внешней
и внутренней
сфер.
выступает в качестве акцептора готовой
электронной пары, предоставляя, свободные
орбитали (5s
и 5p)
для координации; молекулы аммиака
выступают в качестве донора электронной
пары. Химическая связь такого вида
называется донорно-акцепторной
или координационной. По своей природе
- это полярная ковалентная связь,
отличие состоит лишь в способе её
образования. Координационные соединения
состоят из внешней
и внутренней
сфер.
[H3N:←□
Ag□←:
NH3]![]() Cl‾
Cl‾
внутренняя сфера внешняя сфера
Различают внутренние сферы
1.Катионного
характера: [Ag(NH)2]![]() ;[Cu(NH3)4]
;[Cu(NH3)4]![]()
2.Анионного характера:
[Co(CN)4]![]() ;
[Fe(CN)6]
;
[Fe(CN)6]![]()
3.Нейтрального характера: [Fe (CO) 5]
Комплексную
частицу, например,
![]() Cr3+(H2O)0
6
Cr3+(H2O)0
6
![]() 3+ образуют:
3+ образуют:
- Центральный атом (ЦА) или, его так же называют комплексообразователь (КО) – это катион хрома Cr3+
- Лиганды (L) - в нашем примере это молекулы воды, которые координируются вокруг ЦА, их количество (6) соответствует значению координационного числа (КЧ = 6).
Заряд комплексного иона равен алгебраической сумме зарядов ЦА и L:
К2+
![]() Pt2+Cl4-
Pt2+Cl4-
![]() 2-;
2-;
![]() Cr3+(H2O)0
6
Cr3+(H2O)0
6
![]() 3+Cl3-
3+Cl3-![]()
В качестве комплексообразователей может быть любой элемент периодической системы, но наибольшую склонность к комплексообразованию имеют d- и f- металлы, некоторые р-элементы.
Координационное число (КЧ) - определяет количество - связей, образуемых центральным атомом с лигандами (табл.№ 16)
Координационное число зависит от многих факторов: RЦА, заряда ЦА, размеров и валентности лигандов, среды в которой протекает реакция комплексообразования.
Таблица №16 Зависимость КЧ от заряда ЦА
|
Заряд ЦА |
КЧ |
Пример |
|
+1 |
2,3,4, но чаще всего 2 |
Ag+, Cu+, Au+ [Ag (NH3) 2] Cl |
|
+2 |
3,4,5,6,7,8, но чаще всего 4 |
Cu2+, Fe2+, Zn2+, Hg2+, Au2+, Pt2+, Pb2+ [Cu (H2O) 4]SO4, Na2[Zn(OH)4], K4 [Fe(CN)6]
|
|
+3 |
4,5,6,8,9,10,12, но чаще всего 6 |
Fe3+, Cr3+, Co3+ , Al3+, K3 [Fe (CN) 6] |
|
+4 |
6,8, |
Pt4+,Pb4+ , K2 [PtCl6] , K4 [PtCl8] |
Лигандами могут быть нейтральные молекулы или анионы, простые или сложные молекулы органических веществ, у которых имеются неподеленные электронные пары.
По числу химических связей, образуемых лигандами с центральным атомом, лиганды можно разделить на:
-
монодентатные : F‾;
Cl‾; I‾;
![]()
![]() ;
;
![]()
![]()
![]() и т.д..
и т.д..
-бидентатные - :SO4²‾ ;:NH2-(CH2)2 –Н2N: - этилендиаммин (ЭДА) .
- полидентатные: :OOCH2C CH2COО:
![]() -(CH2)2-
-(CH2)2-![]()
:OOCH2С CH2COO:
Этилендиаминтетрауксусная кислота
По наличию заряда, выделяют лиганды:
-анионного типа: CN‾; Cl‾ ; OH‾ ; CNS‾
-нейтрального типа: NH3; H2О ; CO ; NO ; ЭДА .
Комплексные соединения по химической природе лигандов делятся на четыре класса.