

6 курс / Неонатология / Проксимальная_спинальная_мышечная_атрофия_5q_Гузева_В_И_,_Иванов
.pdfБИБЛИОТЕКА ПЕДИАТРИЧЕСКОГО УНИВЕРСИТЕТА
ПРОКСИМАЛЬНАЯ
СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ 5q
Санкт-Петербург
0

Министерство
здравоохранения Российской Федерации
Санкт-Петербургский Государственный Педиатрический Медицинский Университет
ПРОКСИМАЛЬНАЯ
СПИНАЛЬНАЯ
МЫШЕЧНАЯ АТРОФИЯ 5q
Методическое пособие |
САНКТ-ПЕТЕРБУРГ |
для врачей |
2021 |
1
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
УДК 616.832-009.5 ББК 56.12
П80
П80 Проксимальная спинальная мышечная атрофия 5q. Методическое пособие для врачей /
В.И. Гузева, Д.О. Иванов, Ю.В. Петренко [и др.]. – СПб.: СПбГПМУ, 2021. – 20 с.
ISBN 978-5-907443-24-2
Методическое пособие предназначено для практических специалистов перинатальных центров, родильных домов, неврологических больниц и отделений, поликлиник, консультативно-диагностических центров; врачей-неврологов, неонатологов, педиатров и других специалистов, а также студентов медицинских вузов, клинических ординаторов, аспирантов. В пособии систематизированы основные представления о проксимальной спинальной мышечной атрофии 5 q с учетом современных нейрофизиологических, генетических, клинических и терапевтических особенностей заболевания. Особое внимание уделено вопросам ранней диагностики в доклинической стадии заболевания, приведен алгоритм диагностической оценки. В учебном пособии использован материал актуальных научных исследований и разработок.
Авторы:
Гузева В.И. – д.м.н., профессор, заведующий кафедрой неврологии, нейрохирургии и медицинской генетики СПбГПМУ; Иванов Д.О. – д.м.н., профессор, зав. кафедрой неонатологии с курсами неврологии и акушерства-
гинекологии ФП и ДПО, ректор СПбГПМУ; Петренко Ю.В. – доцент, к.м.н., главный внештатный специалист неонатолог СЗФО РФ
Охрим И.В. – к.м.н., доцент кафедры неврологии, нейрохирургии и медицинской генетики СПбГПМУ; Гузева О.В. – д.м.н., доцент, профессор кафедры неврологии, нейрохирургии и медицинской генетики СПбГПМУ; Гузева В.В. – д.м.н., доцент, профессор кафедры неврологии, нейрохирургии и медицинской генетики СПбГПМУ.
Рецензенты:
Скрипченко Н.В. – д.м.н., профессор, зав.кафедрой инфекционных заболеваний у детей ФП и ДПО ФГБОУ ВО СПбГПМУ МЗ России Чутко Л.С. – руководитель центра Поведенческой неврологии, заведующий лабораторией коррекции
психического развития и адаптации Института мозга человека РАН, доктор медицинских наук, профессор, врач-невролог высшей категории, психотерапевт.
УДК 616.832-009.5 ББК 56.12
Утверждено учебно-методическим советом Федерального государственного бюджетного образовательного учреждения «Санкт-Петербургский государственный педиатрический медицинский университет» Министерства здравоохранения Российской Федерации
Выпускается при поддержке Фонда научно-образовательных инициатив «Здоровые дети – будущее страны»
ISBN 978-5-907443-24-2 |
© СПбГПМУ, 2021 |
2
|
СОДЕРЖАНИЕ |
|
1. |
Введение …………………………………………………………………… |
4 |
2. |
Общие сведения ............................................................................................ |
4 |
3. |
Этиология и патогенез .................................................................................. |
5 |
4. |
Классификация……………………………………………………………... |
7 |
5. |
Клинические проявления ............................................................................. |
8 |
6. |
Диагностика ................................................................................................... |
11 |
7. |
Дифференциальный диагноз………………………………………………. |
13 |
8. |
Лечение .......................................................................................................... |
14 |
9. |
Профилактика, уход………………………………………………………... |
17 |
Список литературы ........................................................................................... |
19 |
|
3
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
1. ВВЕДЕНИЕ
Спинальные мышечные атрофии (СМА; англ. spinal muscular atrophy, SMA) составляют группу клинически и генетически гетерогенных наследственных заболеваний с различными типами наследования, характеризующихся прогрессирующими симптомами вялого паралича и мышечной атрофии вследствие дегенерации α-мотонейронов передних рогов спинного мозга.
Наиболее часто встречающаяся форма СМА у детей — проксимальная спинальная мышечная атрофия 5q с аутосомно-рецессивным типом наследования. Заболевание связано с мутациями в генах SMN1 и SMN2, кодирующих белок, участвующий в синтезе сплайсосомы. Выделяют 4 клинических типа проксимальной спинальной мышечной атрофии 5q.
Первые научные описания СМА 1 типа с доказательствами морфологической ее сущности были сделаны в конце XIX в. немецкими неврологами Guido Werdnig (1844–1919) и Johann Hoffmann (1857–1919), впоследствие названная именами ученых.
В 1956 г. шведскими неврологами Е. Kugelberg и L. Welander выделена более благоприятная клиническая форма – СМА 3 типа, имеющая ту же генетическую причину. В дальнейшем выявлены и описаны иные формы СМА, а благодаря совершенствованию генетических исследований в настоящее время подтверждается диагноз и создаются новые генные методы лечения.
Отечественные ученые, сотрудники кафедры и клиники неврологии ФГБОУ ВО СПбГПМУ (ЛПМИ, СПбГПМА), в частности проф. Е.Ф. Давиденкова, проф. Е.А. Савельева-Васильева внесли большой вклад в изучение заболевания, наблюдение и лечение пациентов. Заведующая кафедрой, Главный внештатный детский невролог МЗ РФ проф. В.И. Гузева в составе мультидисциплинарной группы участвовала в подготовке Клинических рекомендаций, утвержденных в 2020 г. и внедрении современных методов лечения СМА в России.
При содействии Правительства РФ организован благотворительный фонд «Круг добра» для поддержки детей с редкими (орфанными) заболеваниями, нуждающихся в дорогостоящем лечении (https://фондкругдобра.рф/). Указ о его создании подписал президент России Владимир Путин 5 января 2021 года. Благодаря фонду дети с этими тяжелыми заболеваниями получают необходимую патогенетическую терапию
Основной задачей практикующих врачей (неонатологов, педиатров и неврологов) является ранняя диагностика заболевания, желательно в доклинической фазе, когда генные препараты являются наиболее эффективными и ребенок сохраняет свои двигательные функции. Именно с этой целью создано данное руководство, где в краткой форме изложены основные сведения о заболевании, ранние клинические симптомы и описаны современные методы лечения.
2. ОБЩИЕ СВЕДЕНИЯ
Проксимальная спинальная мышечная атрофия 5q (СМА) — группа редких (орфанных), клинически гетерогенных наследственных нервно-мышечных за-
4
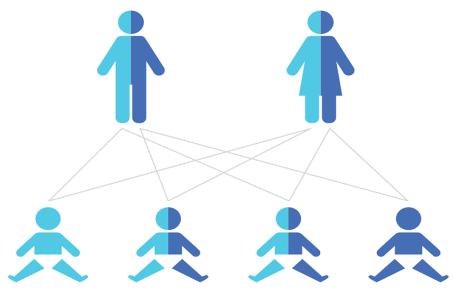
болеваний, вызванных прогрессирующей дегенерацией α-мотонейронов передних рогов спинного мозга, с аутосомно-рецессивным типом наследования (рисунок 1).
Рис. 1. Схема аутосомно-рецессивного типа наследования
Риск рождения больного ребенка в семье, где оба родителя являются носителями патологического гена составляет 25%, вероятность рождения здорового ребенка также составляет 25%. В 50% случаев дети являются носителями рецессивного патологического гена.
Начало заболевания варьирует от рождения до взрослого возраста. Распространенность СМА составляет 1 на 6—10 тысяч новорожденных. Час-
тота носительства заболевания определяется как 1/40–1/50 в популяции в целом.
Дегенерация α-мотонейронов приводит к нарушению работы поперечнополосатой мускулатуры и формированию периферических парезов.
Для СМА характерно отсутствие нарушений чувствительности и интеллек- туально-мнестических функций.
МКБ ШИФРЫ
G 12 Спинальная мышечная атрофия и родственные синдромы.
G 12.0 Детская спинальная мышечная атрофия, I тип (Верднига— Гоффмана).
G 12.1 Другие наследственные спинальные мышечные атрофии.
3. ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Развитие СМА I-IV типов обусловлено недостаточной выработкой белка выживаемости мотонейронов (SMN, survival motor neuron). Белок SMN функционирует в ядре и цитоплазме клеток органов, участвуя в сплайсинге прерРНК, биогенезе малых ядерных рибонуклеопротеинов (мяРНК), в генной экс-
5
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
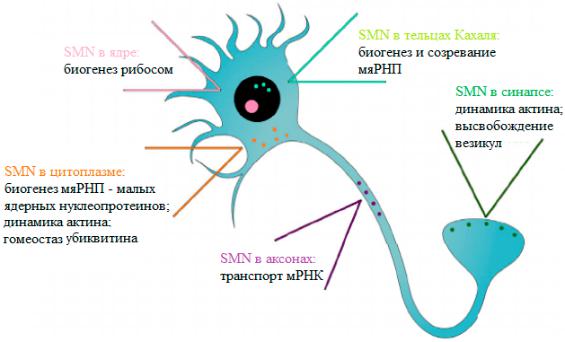
прессии на уровне транскрипции и аксональном транспорте мРНК, в том числе в α-мотонейронах спинного мозга (рисунок 2).
Рис. 2. Функции белка SMN в нейроне
[Источник: Melissa Bowerman, Catherina G. Becker, Rafael J. Yáñez-Muñoz, Ke Ning, Matthew J. A. Wood, et. al.. (2017). Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model. Mech.. 10, 943-954]
Аксоны α-мотонейронов имеют значительную протяженность, поэтому нарушение синтеза белка SMN имеет более драматические последствия для двигательных функций организма.
Уменьшение уровня SMN-белка приводит к аксональным дефектам двигательных нейронов и нарушениям в нервно-мышечных синапсах (рисунок 3). При этом происходит прогрессирующая дегенерация мотонейронов в передних рогах спинного мозга, что проявляется атрофиями и слабостью проксимальных мышц конечностей.
Различают два одноименных гена, кодирующих SMN-белок.
Ген SMN1 расположен в локусе 5q12.2-q13.3. В 95% случаев патология обусловлена делециями экзонов 7 и/или 8 в гене SMN 1. Остальные 5 % являются гетерозиготами по делеции в одной копии гена SMN1 и точковой мутации в другой копии.
Ген SMN2 является функционально неполноценной центромерной копией гена SMN1и продуцирует полноразмерный белок в относительно малых количествах (до 10%), поэтому его изолированная мутация не может являться причиной СМА.
Приблизительно у 80% людей в популяции наблюдается 1-2 копии гена SMN2. Увеличенное число копий гена SMN2 у пациентов с СМА, как правило, повышает выработку функционального белка, что определяет более старший возраст дебюта и менее выраженные клинические проявления заболевания. Од-
6
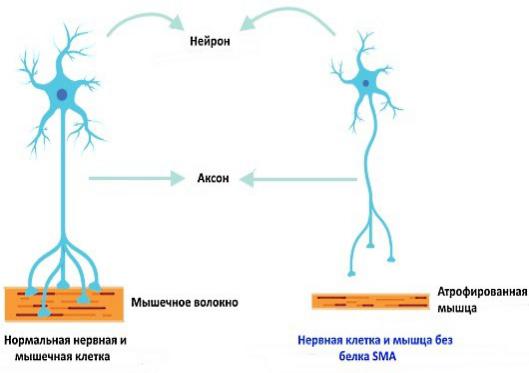
нако, установление типа СМА проводится врачом на основании всей совокупной информации о заболевании у конкретного пациента, независимо от числа копий гена SMN2.
Рис. 3. Механизм развития атрофии мышц в результате повреждения мотонейронов спинного мозга.
[Источник: What is spinal muscular atrophy (SMA)? https://www.togetherinsma.com/en_us/home/introduction-to-sma/smn1-gene.html]
4.КЛАССИФИКАЦИЯ
Вклинической классификации проксимальных спинальных атрофий выделяют 4 типа, в зависимости от возраста дебюта, выраженности клинических проявлений и продолжительности жизни (таблица 1).
Таблица 1
Клиническая классификация типов проксимальных мышечный атрофий 5q
|
|
|
Продолжитель- |
|
|
Возраст |
Двигательные навыки, |
ность жизни |
|
Тип СМА |
дебюта |
при естественном |
||
функциональные возможности |
||||
|
заболевания |
течении |
||
|
|
|||
|
|
|
заболевания |
|
|
|
|
|
|
Тип 1 |
0–6 месяцев |
Голову не держит, не переворачи- |
До 2-х лет |
|
Болезнь Верднига– |
|
вается, не сидит |
|
|
Гофмана |
|
|
|
|
Тип 2 |
6–18 месяцев |
Способность сидеть без поддерж- |
Более 2-х лет |
|
Болезнь Дубовица |
|
ки |
|
|
|
|
|
|
|
|
|
7 |
|
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
|
|
|
|
|
Продолжитель- |
||
|
Возраст |
Двигательные навыки, |
ность жизни |
||||
Тип СМА |
дебюта |
при естественном |
|||||
функциональные возможности |
|||||||
|
заболевания |
течении |
|||||
|
|
|
|
||||
|
|
|
|
|
заболевания |
||
|
|
|
|
|
|||
Тип 3 |
Старше 18 ме- |
Способность стоять и ходить без |
70% |
больных до- |
|||
Болезнь Кугельбер- |
сяцев |
поддержки, |
но |
могут утратить |
живают до 25 лет |
||
га–Веландер |
|
способность |
к самостоятельному |
|
|
||
|
|
передвижению |
|
|
|
||
Тип 4 Поздний тип |
В подростковом |
Передвигаются |
самостоятельно |
10–40 |
лет после |
||
|
или взрослом |
или с поддержкой |
|
манифестации |
|||
|
возрасте |
|
|
|
|
|
|
5. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
При всех типах СМА в разной степени выраженности наблюдаются:
двигательные нарушения в виде периферических парезов и параличей;
симптомы поражения дыхательной системы проявляющиеся гиповентиляцией во сне (апноэ) и бодрствовании;
кардиологическая патология (аритмии, кардиомиопатия и др.);
бульбарные расстройства в виде нарушения глотания, звучности голоса, речи, снижение глоточного, небного рефлексов;
симптомы поражения костной системы. Формируются скелетные деформации, искривление позвоночника с пониженным мышечным тонусом,
контрактуры суставов конечностей.
СМА 1 типа (болезнь Верднига–Гоффмана) характеризуется дебютом до
6 месяцев. Внутриутробно может отмечаться слабое шевеление плода. Основными клиническими признаками СМА 1 типа являются проксималь-
ная, симметричная мышечная слабость, мышечная гипотония и отсутствие моторного развития.
При рождении заболевание может проявляться симптомами:
Слабый и тихий, тонкий «писклявый» крик;
Атрофия и фибрилляции языка;
Бульбарные нарушения (поперхивание, ослабление сосательного, глоточного и кашлевого рефлексов), частые поперхивания, увеличение длительности приема пищи;
Тремор дистальных отделов конечностей;
Синдромокомплекс «вялого ребенка». Диффузная мышечная гипотония до степени атонии, проявляющиеся распластанной «позой лягушки», отсутствием мышечного сопротивления при пассивных движениях, отсутствием или угасанием сухожильных рефлексов (рисунок 4);
Контрактуры челюстного сустава, костные деформации грудной клетки, контрактуры крупных суставов конечностей.
Безусловные рефлексы новорожденных ослаблены или отсутствуют.
8
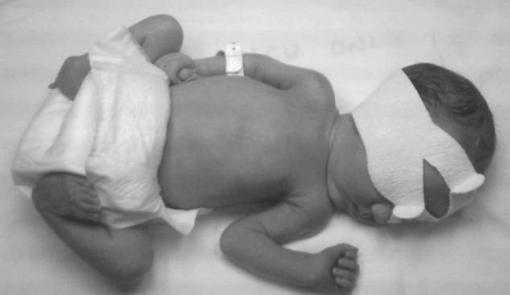
Рис. 4. Ребенок с диффузной мышечной гипотонией, контрактурами суставов.
При прогрессировании заболевания нарастают бульбарные нарушения, дыхательная недостаточность, аспирационный синдром, присоединяются вторичные инфекции. Нарушения дыхания – основная причина возникновения осложнений и смертности пациентов со СМА.
При сборе анамнеза уточняются жалобы на частые пробуждения, наличие храпа, утренние головные боли, сонливость в течение дня для ранней диагностики гиповентиляции и апноэ. Важными являются сведения о частоте инфекционных заболеваний дыхательной системы.
Для выявления бульбарных расстройств тщательно собираются сведения о нарушениях глотания жидкой и твердой пищи, частоту и время поперхиваний (до, во время или после глотания), а также изменениях звучности голоса, наличии охриплости после акта глотания.
Ребенок не осваивает моторные навыки: не держит голову, не переворачивается, не садится. Мышечная слабость достигает степени паралича, полной обездвиженности. Могут сохраняться слабые движения в дистальных отделах конечностей в ответ на прикосновение. Наблюдаются запрокидывание головы, отрицательный верхний рефлекс Ландау – симптом «свешенного белья» (рисунок 5).
Без лечения и должного ухода продолжительность жизни составляет около 2 лет.
СМА 2 типа (болезнь Дубовица). Проявления болезни возникают в 6–18 месяцев, когда сформирована способность удерживать голову самостоятельно и садиться.
Ранними признаками заболевания могут являться:
Фибриллярные подергивания языка;
Бульбарные нарушения (поперхивание, ослабление глоточного и кашлевого рефлексов, слабый и тихий голос);
Мышечная слабость конечностей, диффузная мышечная гипотония, снижение сухожильных рефлексов или арефлексия;
Тремор рук
9
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/