

моноклональных антител, в частности, против CD52 – кампат, а также анти-CD20 - ритуксимаб (мабтера).
Особенности лечения ВКЛ:
1.Пуриновые аналоги (пентостатин, кладрибин) позволяют достичь ремиссии у 60-90% пациентов, при этом 5-летняя выживаемость составляет 90%.
2.Альфа-интерферон способствует развитию положительного эффекта у 80% пациентов, при этом у 10% развивается полная ремиссия.
3.Среди новых подходов (особенно при резистентных формах) – использование моноклональных антител (мабтера) против антигенов, расположенных на опухолевых клетках.
4.Спленэктомия позволяет ликвидировать проявления гиперспленизма и уменьшает объем опухолевого клона.
Прогноз. К прогностическим факторам относят различные специфические хромосомные нарушения:
1)Больные с 13q14 поломкой склонны к более доброкачественному течению заболевания и имеют, как правило, обычную продолжительность жизни.
2)Делеция 11q23 наблюдается у 18% больных и ассоциируется с массивной лимфаденопатией и агрессивным течением лейкемического процесса.
3)Трисомия 12q встречается у 16% больных и связана с атипичной морфологией клеток и плохим исходом заболевания.
4)Цитогенетические поломки 11qили 17qсвидетельствуют о неблагоприятном
прогнозе Неблагоприятными прогностическими факторами являются также
1)мужской пол,
2)лимфоцитоз более 50х109 в дебюте заболевания,
3)содержание более 5% пролимфоцитов в крови,
4)удвоение количества лимфоцитов менее чем за 12 мес,
5)повышение уровня β2-микроглобулина в крови,
6)повышение уровня лактатдегидрогеназы в сыворотке крови,
7)слабый ответ на терапию.
Миелопролиферативные заболевания
- это группа патологических процессов, для которых характерна аномальная пролиферация по крайней мере одной линии гемопоэтических клеток или соединительнотканных структур костного мозга.
Кчислу миелопролиферативных заболеваний относят хронический
миелолейкоз, хронический идиопатический миелофиброз, истинную полицитемию и эссенциальную тромбоцитемию. Хотя группа миелопролиферативных заболевания имеет ряд общих признаков, все же для каждой формы характерно определенное сочетание клинических проявлений, особенностей течения и лабораторных данных. Объединяет все эти заболевания тот факт, что причиной их развития является аномальное размножение клона на уровне полипотентных стволовых клеток, ведущее к пролиферации клеток эритропоэза, гранулоцитопоэза и мегакариоцитопоэза, выраженной в разной степени.
Хронический миелолейкоз
Определение. Хронический миелолейкоз (ХМЛ) - это клональное опухолевое заболевание, вызванное злокачественным перерождением полипотентной стволовой клетки –
93
предшественницы миелопоэза, общей для гранулоцитарного, эритроидного и мегакариоцитарного ростков кроветворения, что обусловливает вовлечение в патологический процесс при этом заболевании всех трех вышеуказанных рядов гемопоэза.
Эпидемиология. Частота ХМЛ составляет 1,5 случая на 100 тыс населения в год. Чаще встречается у мужчин (2 случая), чем у женщин (1,2 случая). Пик заболеваемости приходится на 50-летний возраст. Средняя продолжительность жизни 3-5 лет.
Этиология. Доказанным фактором риска возникновения заболевания считается ионизирующая радиация и некоторые химические агенты (бензол).
Патогенез. Ведущим патогенетическим механизмом ХМЛ является транслокация хромосом (9;22), которая возникает вследствие соматической мутации в стволовой кроветворной клетке. Эта хромосома получила название Филадельфийская по городу, в котором она была обнаружена. Эта транслокация является результатом слияния breakpoint cluster region (BCR) гена хромосомы 22q с ABL (abelson murine leukemia virus) геном, локализованным на хромосоме 9q34. В результате транслокации на 22-й хромосоме появляется новый ген BCRABL, который кодирует слитный белок р210, являющийся тирозинкиназой, определяющей развитие ХМЛ.
Филадельфийская хромосома определяется у 95% больных. Примерно у 5% филадельфийская хромосома является результатом атипичных транслокаций (9;22). В настоящее время можно считать установленным факт, что истинного Ph-негативного ХМЛ не существует. Установлено также, что у больных ХМЛ одновременно сосуществуют Phпозитивные и Ph-негативные, т.е. нормальные стволовые клетки.
Клиника. В клиническом течении ХМЛ выделяют три фазы: хроническую, прогрессирующую (фазу акселерации) и бластный криз. Клинически начальную стадию болезни определить не удается. При бессимптомном течении болезни диагноз выясняется случайно, при выполнении анализа крови.
Главным клиническим признаком болезни в хронической фазе является увеличение размеров селезенки и печени, которые являются следствием появления очагов экстрамедуллярного кроветворения. С увеличением селезенки может быть связано развитие гиперметаболического состояния - лихорадки, потливости, потери массы тела и хронической усталости.
Согласно классификации ВОЗ, верификация прогрессирующей фазы (фаза акселерации) ХМЛ основывается на выявлении одного из следующих признаков:
•нарастающая спленомегалия и рост числа лейкоцитов, не отвечающие на
лечение;
•содержание бластных клеток в крови или костном мозге 10-19%;
•промиелоцитов более 30%
•содержание базофилов в периферической крови не менее 20%;
•персистирующая тромбоцитопения (<100х109/л) или тромбоцитоз (>1000х109/л), не поддающиеся терапии;
•обнаружение дополнительных хромосомных аномалий таких как трисомия 8, 9, 19, 21, делеция Y хромосомы.
Проявлением бластного криза ХМЛ считаются
•увеличение количества бластных клеток в периферической крови или костном мозге до 20% и более (рис. 5.5.),
•выявление экстрамедуллярных бластных пролифератов различных органах и
тканях.
94

С клинических позиций представляется целесообразным выделение лимфоидного и нелимфоидного бластного криза.
Рис. 5.5. Увеличение числа бластных клеток в костном мозге больного хроническим миелолейкозом в фазе бластного криза
Дополнительные методы исследования.
I. Общий клинический анализ крови: нейтрофильный лейкоцитоз со сдвигом до промежуточных форм миелоидного ряда (миелобластов, промиелоцитов), повышение содержания базофилов и эозинофилов (так называемая «эозинофильно-базофильная ассоциация» считается специфическим признаком ХМЛ). Базофилы могут продуцировать гистамин, что может приводить к появлению зуда, диареи, потливости.
Содержание гемоглобина и числа эритроцитов в хронической фазе болезни долгое время остается в норме. Наоборот, в этой фазе исходно может выявляться эритроцитоз с повышением содержания гемоглобина. Количество тромбоцитов у больных в хронической фазе также может быть в норме или даже увеличено (рис. 5.4.). В фазе бластного криза наблюдаются анемия и тромбоцитопения.
Рис. 5.4. Тромбоцитоз в крови больного хроническим миелолейкозом
II. Миелограмма: в хронической фазе ХМЛ отмечается повышение клеточности в основном за счет незрелых форм гранулоцитов, абсолютное количество мегакариоцитов у большинства больных превышает норму.
III. Биохимический анализ крови: понижение активности щелочной фосфатазы нейтрофилов.
IV. Цитогенетическое исследование: обнаружение Филадельфийской хромосомы (Phхромосомы).
V. Молекулярногенетическое исследование: выявление патологического гена BCR-
ABL.
95
Диагноз. Диагноз устанавливается на основании обнаружения при цитогенетическом исследовании Ph-хромосомы и при молекулярногенетическом исследовании гена BCR-ABL.
Дифференциальный диагноз необходимо проводить с другими миелопролиферативными заболеваниями. Главными дифференциально-диагностическими признаками являются наличие Ph-хромосомы и гена BCR-ABL.
Лечение. Задачами терапии ХМЛ является
-стабилизация клинического течения,
-нормализация показателей крови,
-достижение цитогенетического ответа (элиминации клеток, содержащих Phхромосому).
1. Пересадка аллогенных гемопоэтических стволовых клеток от HLA-совместимого донора является терапией выбора для пациентов моложе 50 лет при достижении большого цитогенетического ответа. Наилучший прогноз наблюдается у пациентов в возрасте моложе 30 лет, которым трансплантация стволовых клеток выполнена в хронической фазе в течение первого года после установления диагноза.
2.Иматиниб (гливек) - первый представитель нового класса препаратов, блокирующий ВСR-ABL-тирозинкиназу. Начальная доза - 400 мг однократно во время еды на ночь. Назначение этого препарата обеспечивает полный гематологический ответ у 96% и полный цитогенетический ответ у 68%. Его назначение на ранних фазах заболевания может позволить выключить лейкемические клетки настолько эффективно, что у них не остается шансов на развитие дополнительных поломок. Однако если лейкозный процесс прогрессирует, в опухолевых клетках появляются другие хромосомные поломки. Эти клетки становятся независимыми от первоначальных онкогенных белков. Вот почему, гливек менее эффективен в фазе акселерации и еще менее результативен при бластном кризе.
3.Альфа-интерферон – позволяет достичь цитогенетического ответа у 18-56%.
4.Гидреа позволяет контролировать уровень лейкоцитов, улучшать показатели лейкоцитарной формулы, уменьшать размеры селезенки в хронической фазе процесса, однако не оказывает влияния на цитогенетические нарушения.
5.Наступление фазы акселерации или бластного криза является следствием перехода
вполиклоновую стадию и требует пересмотра терапевтической тактики. Пациентов лечат по протоколам для терапии острых лейкозов.
Прогноз. Прогностически неблагоприятными признаками ХМЛ считаются:
1)возраст в момент установления диагноза 60 лет и более;
2)число бластных клеток в крови 3% и более или в костном мозге 5% и более;
3)число базофилов в крови – 7% и более или в костном мозге 3% и более;
4)число тромбоцитов более 700 тыс в мкл.
Хронический идиопатический миелофиброз
Определение. Хронический идиопатический миелофиброз (МФ) или сублейкемический миелоз, миелоидная метаплазия, алейкемический миелоз – это клональное опухолевое заболевание, морфологическим субстратом которого является полипотентная гемопоэтическая стволовая клетка – предшественницы миелопоэза с вовлечением в процесс гранулоцитарного, эритроидного и мегакариоцитарного ростков, сопровождающееся развитием фиброза костного мозга.
96
Эпидемиология. МФ обнаруживается преимущественно у лиц пожилого и старческого возраста, чаще у мужчин, чем у женщин. Распространенность его приблизительно составляет 0,5-1,5 случаев на 100 000 населения.
Этиология и патогенез. Причины возникновения МФ не до конца выяснены. В костном мозге больных происходит развитие фиброза, причем наиболее интенсивно – в местах скопления мегакариоцитов. Установлено, что мегакариоциты синтезируют в большом количестве фактора роста, которые стимулируют деятельность фибробластов и синтез коллагена. Степень фиброза коррелирует с количеством диспластических мегакариоцитов, поэтому именно гиперплазией мегакариоцитов и их повышенной деструкцией объясняется интенсивное накопление в костном мозге факторов роста, стимулирующих образование фиброзной ткани. Одновременно наблюдается повышенное образование сосудов костного мозга, что также усиливает фиброзно-остеосклеротические изменения.
Клиника. Увеличение размеров селезенки является ведущим клиническим проявлением МФ. Иногда появляются выраженные боли в животе, обусловленные развитием инфарктов селезенки и периспленита. Причинами увеличения селезенки является миелоидная метаплазия, портальная гипертензия, а также усиление ее депонирующей и секвестрирующей функций.
Гепатомегалия обнаруживается у 50% пациентов. Нередким симптомом является мочекислый диатез, образование уратных камней в мочевых путях с картиной мочекаменной болезни.
Дополнительные методы исследования
I. Общий клинический анализ крови: а) анемия - вследствие
-замещения кроветворного костного мозга соединительной тканью,
-гиперспленизма,
-гемолиза эритроцитов (из-за усиления секвестрирующей способности селезенки),
б) у некоторых больных заболевание дебютирует эритремической фазой, сопровождающейся повышением уровня эритроцитов и гемоглобина.
в) количество лейкоцитов нормальное или умеренно увеличенное. При прогрессировании патологического процесса развивается дейкопения.
г) в лейкоцитарной формуле – нейтрофилез со сдвигом влево, вплоть до промиелоцитов и миелобластов.
д) вовлечение в патологический процесс мегакариоцитарного ростка сопровождается значительным тромбоцитозом, а также появлением в периферической крови осколков мегакариоцитов и гигантских форм тромбоцитов. При прогрессировании патологического процесса развивается тромбоцитопения.
II. Миелограмма: пункция грудины из-за выраженного фиброза нередко оказывается неудачной, аспират сухой и скудный.
III. Трепанобиопсия. Убедительным признаком МФ является обнаружение гиперплазии всех ростков миелоидного кроветворения – гранулоцитарного, эритроидного и мегакариоцитарного с очагами фиброза при трепанобиопсии костного мозга (рис. 5.6.). При этом выделяют следующие морфологические стадии заболевания: I стадия – неравномерная пролиферация клеток гранулоцитарного, эритроидного и особенно мегакариоцитарного ростка с очагами ретикулинового миелофиброза, II стадия – очаги грубоволокнистого коллагенового миелофиброза, нарушающего архитектонику костного мозга, в тяжах соединительной ткани сохраняются островки гемопоэза с преобладанем мегакариоцитов, IIIIV стадии – присоединение остеомиелосклероза.
97
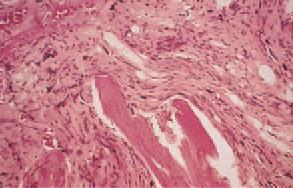
Рис. 5.6. Морфологическая картина биоптата костного мозга при идиопатическом миелофиброзе.
Диагноз. Критериями установления диагноза являются:
1.Спленомегалия, обусловленная миелоидной метаплазией.
2.Коллагеновый миелофиброз, выявляемый при гистоморфологическом исследовании костного мозга и занимающий более половины препарата.
3.Лейкоэритробластическая картина периферической крови с наличием каплевидных эритроцитов.
4.Отсутствие заболеваний, которые могут быть причиной развития миелофиброза (см. ниже).
Дифференциальный диагноз. Необходимость дифференциальной диагностики МФ возникает с ХМЛ и истинной полицитемией. При МФ в отличие от ХМЛ не обнаруживается Phхромосома и BCR-ABL ген. При истинной полицитемии при трепанобиопсии отсутствует фиброз костного мозга.
Прежде чем выставлять диагноз идиопатического миелофиброза, необходимо исключить все возможные причины для развития вторичного МФ:
1.Злокачественные новообразования
2.Инфекции
3.Ходжкинские и неходжкинские лимфомы
4.Острый лейкоз
5.ВКЛ
6.Множественная миелома
7.ХМЛ
8.Истинная полицитемия
9.СКВ
10.Почечные остеодистрофии
11.ВИЧ
12.Гиперпаратиреоз
Лечение.
1.Гидреа – снижает число лейкоцитов и выраженность органомегалии.
2.Альфа-интерферон – снижает число лейкоцитов и тромбоцитов
3.Преднизолон – при гемолитической анемии, тромбоцитопении, спленомегалии, клеточном гиперметаболизме.
4.Аллопуринол при мочекислом диатезе
5.При анемии – трансфузии эритроцитарной массы
98