

metodNasledPat08
.pdfинтерстиции почек, лимфоузлах, селезѐнке, печени, слизистой ЖКТ, костном мозге, оболочках глаза выявляются кристаллы цистина прямоугольной или гексагональной формы.
Течение болезни тяжѐлое. Смерть от хронической почечной недостаточности в возрасте не старше 10 лет.
Пациенты с ювенильной формой живут до 30 лет. Величины цистина в лейкоцитах повышены в 30–50 раз. Кроме почек, поражаются суставы.
Взрослая форма – самая благоприятная, патологический процесс протекает медленно, уровень цистина в лейкоцитах не больше 500–600% от нормы. Почки практически не страдают, наблюдаются фотофобия, головные боли, слѐзотечение.
Для установки диагноза используют фибробласты из культуры тканей или лейкоциты, где обнаруживается накопление свободного цистина.
Лечение в основном симптоматическое. При детской и юношеской формах помогает трансплантация почек.
Недостаточность сульфитоксидазы – наследственное заболевание
(аутосомно-рецессивный тип), описаны редкие случаи. На последних этапах катаболизма цистеина (схема 9), когда окисляется его сульфгидрильная группа, используется сульфитоксидаза, преобразующая сульфит в сульфат, обычно экскретируемый с мочой. Его отсутствие в данной биологической жидкости свидетельствует о блоке этого фермента.
Продолжительность жизни больных с подобной патологией не достигает 3-х лет; основные симптомы: поражение центральной и вегетативной нервных систем (низкий IQ, судороги, атаксия, гемипарез и т.д.). В моче повышено содержание сульфитов.
Терапия не разработана.
5.2. Генные повреждения в метаболизме нуклеотидов
Мононуклеотиды выполняют не только функции макроэргов (АТФ, ГТФ, ЦТФ, УТФ), служат вторичными посредниками (цАМФ, цГМФ), коферментами (ФМН), они являются мономерами в генезе полинуклеотидов (ДНК, РНК). В настоящее время считают, что специфический компонент мононуклеотида – пуриновое или пиримидиновое основание – в клетках организма человека имеет только эндогенное происхождение (синтез de novo). Правда, при определѐнных обстоятельствах (у эмбриона, в регенерирующих тканях, опухолях) возможно использование готовых оснований или нуклеозидов, получившихся при катаболизме полинуклеотидов («путь спасения»). Одним из ключевых ферментов последнего процесса служит гипоксантингуанинфосфорибозилтрансфераза (ГГФРТ) (схема 10).
Если повреждается аминокислотная последовательность в цепи этого энзима, то резко нарушается метаболизм пуриновых нуклеотидов у плода. При этом характерно наследование по рецессивно-сцепленному с Х-
хромосомой типу (синдром Лѐша – Найхана, syndrome Lesch-Nyhan),
поэтому болеют только лица мужского пола. У рождѐнных фенотипически
71
здоровыми через несколько месяцев жизни возникает задержка физического развития, позднее присоединяются гиперрефлексия, судороги. Особенно опасно аутоагрессивное поведение: дети начинают кусать, жевать свои пальцы, губы, слизистую щѐк, что приводит к увечьям. Коэффициент интеллектуальности не достигает 30.
Угнетение активности гипоксантингуанинфосфорибозилтрансферазы тормозит использование гуанина в реакциях синтеза нуклеотидов и он преобразуется в мочевую кислоту (схема 10). Для таких больных характерна гиперурикемия, провоцирующая подагрические отложения в суставах.
ДНК |
дГТФ |
|
дАТФ |
ДНК |
РНК |
ГТФ |
Синтез de novo |
АТФ |
РНК |
|
ГМФ |
Инозинмонофосфат |
АМФ |
|
ГГФРТФ-аза
Синдром Лѐша - Найхана
Гуанозин
ФРПФ |
Инозин |
Аденозин |
|
|
Аденозин- |
Гуанин |
|
дезаминаза |
Недостаточность
аденозиндезаминазы
Гипоксантин
КсантинДГ
Ксантинурия
Ксантин
КсантинДГ
Мочевая кислота
Моча
Схема 10. Обмен пуринов в норме и при патологии.
72
Для диагностики используют эритроциты или фибробласты, в них исследуют активность энзима; у больных синдромом Лѐша – Найхана она отсутствует.
Попытки терапии не достаточно эффективны: с помощью диетотерапии (исключение из рациона продуктов, богатых нуклеотидами), препаратов, способствующих выведению мочевой кислоты, добиваются снижения гиперурикемии, выраженности подагрических артритов, но неврологическая симптоматика остаѐтся на прежнем уровне.
Описаны также блоки практически всех ферментов катаболизма пуринов,
одним из которых является недостаточность аденозиндезаминазы (схема
10). Наследуется по аутосомно-рецессивному типу. Очень редкое заболевание, представляет тяжѐлую комбинированную иммунную недостаточность, для которой характерны нарушения и клеточного, и гуморального иммунитетов. Жизнь ребѐнка сохраняется только с помощью специальных скафандров, отделяющих организм от внешних факторов среды.
Эта болезнь первая из наследственных, для терапии которой была использована генная инженерия: введѐн здоровый ген и у больного ребѐнка исчезла необходимость находиться в стерильных условиях (гл. 9).
Ксантинурия (схема 10) - редкая наследственная патология, в основе которой лежит дефект ксантиндегидрогеназы печени и слизистой кишечника. Аутосомно-рецессивный тип наследования. Количество непреобразованного в мочевую кислоту ксантина в крови увеличивается, больше его экскретируется почками. Но так как он ещѐ хуже, чем ураты растворим в воде, - с возрастом образуются ксантиновые камни, они рентген неконтрастны, что затрудняет диагностику. Заболевание протекает благоприятно. Больные иногда жалуются на боли в мышцах после физической нагрузки из-за отложения кристаллов ксантина.
Диагноз ставится на основании гипоурикемии, гиперксантинемии, ксантинурии, а также после изучения активности ксантиндегидрогеназы в
биоптате тощей кишки пациента.
Лечение в основном симптоматическое. Гомозиготы должны ограничивать потребление пуринов, пить больше жидкости.
Оротацидурия
Что касается метаболизма пиримидиновых нуклеотидов, то в настоящее время известно лишь одно заболевание, причиной которого служит генетическое повреждение в синтезе УМФ.
В настоящее время установлено, что ключевые ферменты синтеза
оротатфосфорибозилтрансфераза и оротидилмонофосфатдекарбоксилаза
– два домена одной полипептидной цепи, мутации в которой обычно сопровождаются угнетением работы обоих энзимов. Встречается довольно редко.
Замедление синтеза пиримидиновых нуклеотидов провоцирует угнетение генеза РНК, ДНК, деление клеток, в первую очередь, снижается скорость
73
гематопоэза. Отсюда кардинальный симптом – мегалобластическая анемия, хотя наблюдается и в целом задержка физического развития.
В постановке диагноза помогает обнаружение повышенных количеств оротовой кислоты в моче, а также сниженная активность дефектных ферментов в гепатоцитах, лейкоцитах, эритороцитах, фибробластах.
В терапии используют введение продуктов угнетѐнных реакций, в основном – уридина.
5.3. Альтерации в синтезе и распаде гема 5.3.1. Порфирии
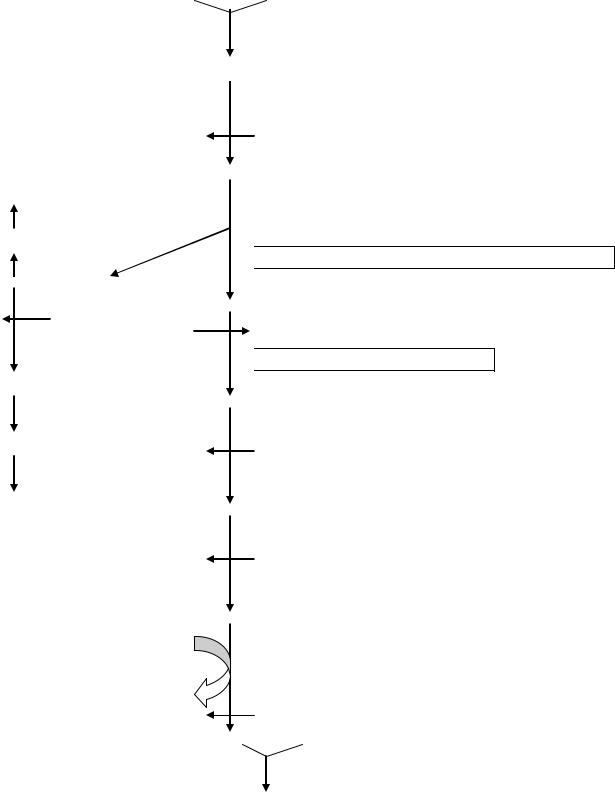
Синтез гема – многостадийный процесс, в котором участвуют несколько ферментов. В настоящее время описаны наследственные заболевания – порфирии (porphyra – пурпурная краска), причинами которых являются мутации, способные вызвать блок любого из энзимов образования данного продукта (схема 11). В зависимости от органной локализации нарушений генеза порфиринов различают эритропоэтические (в костном мозге) и печѐночные (в гепатоцитах). Преобладает аутосомно-доминантный тип наследования.
Подобные недуги встречаются очень редко, несколько чаще диагностируются эритропоэтические порфирии (например,
эритропоэтическая протопорфирия вследствие блока феррохелатазы). Из-за неэффективности синтеза гема в клетках эритроидного ряда накапливаются его предшественники, стимулирующие СРО и тем самым провоцирующие гемолиз (развитие гемолитической анемии, желтухи).
В плазме крови повышаются концентрации уропорфириногена, копропорфириногена, которые в нижних отделах мочевыделительной системы окисляются кислородом и в таком виде выделяются с мочой. Обладая сродством к белкам кожи и соединительной ткани, порфирины задерживаются в них, часто стимулируя сдвиги в окраске (красный цвет зубов). Их накопление в эпителии увеличивает чувствительность кожи к УФО, следствием чего могут быть язвы, превращающиеся затем в грубые шрамы. Высок риск их инфицирования с переходом воспаления на хрящевую и костную ткани. Конечности деформируются, возможна тугоподвижность суставов (контрактуры). Для печѐночных порфирий более характерны неврологические нарушения (боли по ходу нервных стволов, клиника «острого живота»).
Специфическая диагностика практически отсутствует, базируется на данных анамнеза, клиники. Обращает на себя внимание изменение цвета мочи (часто красно-коричневого) за счѐт необычно больших количеств
уропорфиринов или копропорфиринов.
Лечение преимущественно симптоматическое. В тяжѐлых случаях (при гемолитических кризах) проводят спленэктомию. Необходима защита от солнечных лучей, рекомендуются специальные кремы, мази; днѐм лучше находиться в затемнѐнном помещении.
74
Прогноз обычно неблагоприятный. Смерть провоцируется тяжѐлыми кожными поражениями, сниженным иммунитетом, интоксикацией, острыми гемолитическими кризами.
Сукцинил КоА + Глицин
 5-аминолевулинатсинтаза
5-аминолевулинатсинтаза
|
5-аминолевулиновая кислота |
|
|
|
Дегидратаза 5-АЛК |
|
|
|
|
|
Порфирия, вызванная дефицитом дегидратазы |
|
|
5-АЛК |
|
Порфобилиноген |
|
Моча |
|
Порфобилиногендезаминаза |
|
|
уропорфириноген-косинтаза |
Уропорфирин I
 Врождѐнная эритропоэтическая порфирия
Врождѐнная эритропоэтическая порфирия
Уропорфириноген I
Уропорфириноген III
Уропорфириногендекарбоксилаза
 Поздняя кожная порфирия
Поздняя кожная порфирия
Копропорфириноген I
Копропорфириноген III
|
Копропорфириногеноксидаза |
|||
Копропорфирин I |
|
|
|
|
|
|
Наследственная копропорфирия |
||
Моча |
Протопорфириноген IX |
|||
|
|
Протопорфириногеноксидаза |
||
|
|
|
|
|
|
|
Порфирия variegata |
|
|
|
Протопорфирин IX |
|||
|
Fe++ |
|||
|
Феррохелатаза |
|||
|
2Н+ |
|||
|
|
Эритропоэтическая порфирия |
|
|
|
Гем + Протеин |
|||
Гемоглобин Схема 11. Система ферментов синтеза гема в норме и при патологии.
75
5.3.2. Наследственные гипербилирубинемии
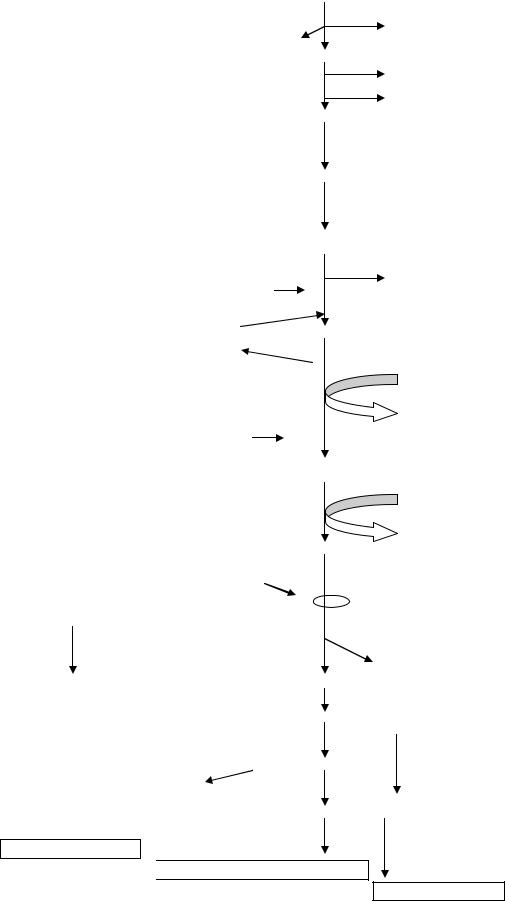
Билирубин – конечный продукт катаболизма гема (схема 12) или предшественников в процессе его неэффективного образования. Отсюда причины накопления этого соединения в плазме крови (гипербилирубинемии) могут носить самый разнообразный характер. В зависимости от локализации нарушений желтухи (следствие увеличенного содержания данного желчного пигмента в эпителии, слизистых из-за его сродства к белкам этих тканей)
делят на гемолитическую, паренхиматозную, механическую. Развитие первых двух форм может быть связано с дефектами структур белков, функции которых состоят в обеспечении нормального метаболизма билирубина (его синтеза и катаболизма), отсюда вероятен их наследственный характер.
Вариации генетических повреждений, провоцирующих усиление гемолиза, следующие:
1)блок того или иного фермента генеза гема порфирии (гл.5.3.1);
2)сдвиги аминокислотной последовательности в протеинах мембран клеток эритроидного ряда, что снижает их резистентность, стимулируя разрушение красных кровяных телец (наследственный микросфероцитоз);
3)развитие подобных изменений в цепях глобина – белкового компонента гемоглобина, что сказывается на его жизнеспособности
(гемоглобиноз S; гл. 5.4.2); блок синтеза целой цепи (талассемии);
4)похожие дефекты в энзимах, ответственных за метаболизм в эритробластах (гликолиз, АРЗ) (см. учебное пособие «Биохимия эритроцитов»).
Иными словами, часть гипербилирубинемий носит вторичный характер, обусловленный нарушением структуры мембран клеток эритроидного ряда или блоком того или иного энзима гликолиза, АРЗ.
Многообразие последствий, способствующих преждевременному распаду эритроцитов, не позволяет описать все заболевания, возникающие при этом. Остановимся на отдельных из них.
Довольно часто встречается болезнь Минковского – Шаффара (morbus Minkowski-Chauffard) (1:4500), наследуется по аутосомно-рецессивному типу. В результате генетического дефекта изменяется аминокислотная последовательность в мембранном белке спектрине, что сказывается на проницаемости плазмолеммы: повышается способность внеклеточных ионов Na+ поступать в цитоплазму эритроцита, принося с собой избыточные количества воды. Эритроидная клетка за счѐт неѐ увеличивается в размерах, принимает форму шара (сферы, отсюда - сфероцитоз), теряет способность к упругой деформации, быстро повреждается, еѐ содержимое оказывается в плазме крови.
Для больных детей характерна следующая триада симптомов: анемия, желтуха, спленомегалия, а также обычно регистрируются дефекты костной ткани («башенный» череп, широкая переносица, аномалии зубов, глазной орбиты, синдактилии).
76
Гемоглобин-гаптоглобин
Красный |
Гемоксигеназа |
|
|
|
|||||||||
костный мозг, |
|
|
|
|
СО |
Гаптоглобин |
|
||||||
печень, |
Вердохромальдегидглобин |
|
|||||||||||
селезѐнка |
|
|
|
|
|
Глобин |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
Железо |
|
|
|
|
|
|
|
|
|
|
|
|
Биливердин |
|
||
|
|
|
|
|
|
Биливердин- |
|
|
|
||||
|
|
|
|
|
|
редуктаза |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
Билирубин |
|
||
|
|
|
|
|
|
|
|
|
|
|
|||
Плазма крови |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
Билирубин + альбумин |
|
|
||||
|
|
|
|
|
|
|
|
|
Альбумин |
|
|||
|
|
|
|
|
Синдром Жильбера |
|
1 |
|
|||||
|
|
|
|
|
|
|
|
||||||
Печень |
Y, Z-лигандины |
|
|
Билирубин + Y, Z-лигандины |
|
||||||||
Гепатоцит |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
УДФГК |
|
|
|
|
|
|
|
|
УДФ-глюкуронил- |
|
|
|
||||
|
|
|
|
|
|
трансфераза |
УДФ |
|
|||||
|
|
|
|
Синдром Криглера-Найяра |
|
2 |
|
|
|
||||
|
|
|
|
|
|
|
Билирубин-моноглюкуронид |
|
|||||
|
|
|
|
|
|
УДФ-глюкуронил- |
УДФГК |
|
|||||
|
|
|
|
|
|
трансфераза |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
УДФ |
|
|
|
|
|
|
|
|
Билирубин-диглюкуронид |
|
||||||
|
|
|
|
Синдром Дабина-Джонсона |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
3 |
|
|
|
||
|
|
|
|
Желчевыводящие пути |
|
|
|
Система транспорта |
|
||||
|
|
|
|
|
|
Бета-глюкуронидаза |
|
|
|
||||
|
|
|
|
|
|
микрофлоры |
Глюкуронат |
|
|||||
|
|
|
|
Тонкий кишечник |
|
|
Билирубин |
|
|||||
|
|
|
|
|
|
|
Редуктазы |
микрофлоры |
|
||||
|
|
|
|
|
|
|
|
|
|
Уробилиногены |
|
||
|
|
|
|
Печень |
Прото- |
i-уробилиноген |
|
||||||
|
|
|
|
|
|
пент- |
|
|
|
|
|
|
|
|
|
|
|
|
|
диапент |
|
|
l-уробилиноген |
|
|||
l-уробилин мочи
 l-уробилин плазмы крови
l-уробилин плазмы крови
l-уробилин кала
Схема 12. Схема распада гемоглобина в норме и при патологии.
77

Высока вероятность развития острого гемолитического криза (гипертермии, одышки, слабости, бледности кожных покровов, тахикардии, болей в животе, печѐночной недостаточности).
Единственным эффективным способом лечения в настоящее время служит спленэктомия, также используют гепатопротекторы, мембраностабилизаторы.
Генетический дефект, провоцирующий синтез аномальных фосфолипидов мембран эритроцитов, проявляется симптомами гемолитической анемии, гемолитической желтухи, ростом в плазме крови уровня гемоглобина. Известен как syndrome Haden, наследуемый по аутосомно-доминантному типу. Встречается редко. Сведения о продолжительности жизни противоречивы.
Примером мутации, приводящей к нарушению метаболических процессов в эритроцитах и провоцирующей их усиленный распад, может служить
наследственная несфероцитарная гемолитическая анемия, описанная в
1968 году.
В еѐ основе лежит аутосомно-рецессивный тип наследования. Причиной является снижение активности глюкозофосфатизомеразы (глюкозо-6-фосфат Е фруктозо-6-фосфат, где Е – данный фермент). Этот вариант анемии занимает третье место по частоте среди подобных патологий (после недостаточности глюкозо-6-фосфатдегидрогеназы, пируваткиназы). Повреждения гликолиза сопровождаются дефицитом энергии, необходимой клетке; еѐ мембраны теряют свою резистентность, ускоряется гемолиз; в результате увеличивается концентрация свободного билирубина, отсюда развивается иктеричность склер, кожных покровов, часты гепато-, спленомегалии. Больных отмечает умеренная умственная отсталость, высока
частота возникновения острых гемолитических кризов.
Диагноз ставится путѐм выявления активности поражѐнного фермента. Из-за малого числа диагностированных больных схема лечения не разработана.
В настоящее время всѐ чаще обнаруживается генетический дефект
глюкозо-6-фосфатдегидрогеназы (Г-6-Ф-ДГ) – энзима, играющего ключевую роль в обеспечении АРЗ эритроцита. Заболевание известно под термином –
недостаточность глюкозо-6-фосфатдегидрогеназы. Носители повреждѐнного гена (до 10% и выше) встречаются чаще в районах, где распространена малярия. Наследуется как сцепленный с Х-хромосомой рецессивный признак.
Все эритроциты мутантного происхождения чувствительны к провоцирующим факторам: конским бобам, голубике, чернике, крыжовнику, чѐрной смородине, папоротнику, некоторым лекарствам (сульфаниламидам, аспирину, амидопирину, викасолу и т.д.), промышленным окислителям. После их поступления в организм развивается усиленный гемолиз, сопровождающийся клиникой гемолитического криза (гипербилирубинемией, желтушностью, слабостью, одышкой, сердцебиением и другими симптомами анемии). Вслед за этим возможна
78
острая почечная недостаточность из-за гемоглобинурии. Последнее явление объясняется тем, что из-за аномалии Г-6-Ф-ДГ снижаются способность восстановления метгемоглобина, устойчивость к действию окислителей, стабильность мембран эритроцитов.
Если в организм не попадают в неадекватных количествах вышеперечисленные вещества, данная патология протекает практически бессимптомно. Но вследствие высокого процента встречаемости и опасности развития тяжѐлого состояния при неблагоприятных условиях эксперты ВОЗ рекомендуют профилактически определять у населения активность фермента в эритроцитах. При обнаружении низкой эффективности его работы следует делать соответствующую пометку в паспорте индивида для своевременного оказания квалифицированной помощи. Оперативное вмешательство – спленэктомия – приносит облегчение больному.
Что касается биохимических изменений в крови, моче и кале, свидетельствующих о нарушениях метаболизма билирубина, они практически однотипны при гемолитических желтухах, независимо от локализации дефекта. Поэтому для уточнения диагноза требуется либо изучение активности специфических ферментов, содержания их субстратов или продуктов, либо выяснение последовательности аминокислот в подозрительных протеинах.
Терапия зависит от места и характера повреждения.
Другой вариант наследственных желтух обусловлен различными генетическими повреждениями белков гепатоцитов. Среди них выделяют
синдромы Жильбера, Криглера-Найяра, Дабина-Джонсона (morbus Gielbert, morbus Crigler-Najjar, morbus Dubin-Johnson) (схема 12).
По мнению различных исследователей, причинами первого заболевания могут быть или дефект гена уридиндифосфатглюкуронилтрансферазы, или,
что более вероятно, точечные мутации, приводящие к изменениям в структурах Y- либо Z-лигандинов - белков, ответственных за захват и перенос билирубина в цитоплазму гепатоцита (схема 12). Наследование по аутосомно-доминантному типу с высокой пенетрантностью. Распространѐнность заболевания в популяции 5 – 7%, мальчики болеют в два раза чаще девочек. Первые симптомы появляются в период полового созревания, они включают непостоянную желтушность кожных покровов, преимущественно под мышками, на шее, за ушами, субиктеричность склер. Окраска может усиливаться под влиянием стрессов, инфекций, физических нагрузок. Гипербилирубинемия невысокая (до 30-80 мкмоль/л, в основном за счѐт свободной формы жѐлчного пигмента) (табл. 9).
Течение благоприятное, на продолжительности жизни не сказывается, правда, у таких больных чаще развиваются поражения ЖКТ, но без повреждений печени. Возможны астеновегетативные нарушения в виде повышенной утомляемости, потливости, эмоциональной лабильности.
Терапия включает щадящий режим, полноценную диету, однако без жирных сортов мяса, копчѐнностей, пряностей, консервов.
79
Синдром Криглера-Найяра (morbus Crigler-Najjar) – одна из наиболее тяжѐлых форм наследственных пигментных гепатозов, обусловленная недостаточностью уридиндифосфоглюкуронилтрансферазы (cхема 12) и
проявляющаяся выраженной гипербилирубинемией (неконъюгированного) (связанная фракция отсутствует), желтухой, возникающей уже в первые сутки после рождения и сохраняющейся на протяжении всей короткой жизни (табл. 9). Встречаются два типа: I – передается по аутосомно-рецессивному, II – по аутосомно-доминантному типам наследования. Концентрация билирубина в крови достигает 300-500 мкмоль/л и даже выше, что в большинстве случаев приводит к развитию ядерной желтухи и энцефалопатии (тоническим и клоническим судорогам, глазодвигательным расстройствам). Частота не установлена.
Диагноз подтверждается определением активности УДФГ-трансферазы в биоптате печени. Моча – светлая; кал, жѐлчь – бесцветные.
Прогноз неблагоприятен, хотя при II типе дефицит энзима несколько менее выражен. С помощью фототерапии пытаются уменьшить уровень гипербилирубинемии, делаются попытки производить пересадки в брюшную полость микроинкапсулированных гепатоцитов. В настоящее время единственный способ лечения – трансплантация печени.
Таблица 9
Основные проявления наследственных паренхиматозных желтух
Синдром |
|
Синдром |
|
|
Синдром |
|
Синдром |
||
Жильбера |
|
Криглера - Найяра |
ДабинаДжонсона |
Ротора |
|
||||
Высокий |
уровень |
Подъѐм |
значений |
Повышение |
коли- |
Рост |
содержания |
||
неконъюгированного |
неконъюгированного |
чества конъюгиро- |
конъюгированно- |
||||||
билирубина в крови |
билирубина |
в |
крови |
ванного билируби- |
го |
(моноглюку- |
|||
|
|
(особенно при типе I) |
на в крови |
|
ронида) |
билиру- |
|||
|
|
|
|
|
|
|
бина в крови |
||
Снижение |
величин |
Уменьшение уровня |
Возрастание |
|
Увеличение цифр |
||||
стеркобилиногена в |
стеркобилиногена в |
общей |
массы |
общих |
копропор- |
||||
крови, моче, кале у |
крови, моче, кале |
неконъюгированно |
фиринов в моче |
||||||
отдельных |
пациен- |
|
|
|
го билирубина в |
|
|
|
|
тов |
|
|
|
|
крови |
|
|
|
|
Увеличение |
содер- |
Значительный |
рост |
Желудочно- |
|
Возможны |
|||
жания моноглюку- |
количества |
|
|
кишечные |
|
желудочно- |
|||
ронида билирубина в |
моноглюкуронидов |
расстройства |
|
кишечные |
|||||
жѐлчи |
|
билирубина в жѐлчи. |
|
|
расстройства |
||||
|
|
Билирубиновая |
|
|
|
|
|
|
|
|
|
энцефалопатия |
(при |
|
|
|
|
|
|
|
|
типе I) |
|
|
|
|
|
|
|
Два клинически сходных редких гепатоза (синдром Ротора, morbus |
|||||||||
Rotor и синдром Дабина-Джонсона, |
morbus Dubin-Johnson) |
развиваются |
|||||||
80