

-
Комбинированные Т- и В- клеточные иммунодефициты.
-
Х- сцепленная тяжелая комбинированная иммунная недостаточность
-
Тяжелая комбинированная иммунная недостаточность с дефицитом аденозиндезаминазы
-
Тяжелая комбинированная иммунная недостаточность с дефицитом пуриннуклеозидфосфорилазы
-
Синдром Оменна
-
Тяжелая комбинированная иммунная недостаточность, обусловленная дефицитом JAK3
-
Дефицит молекул главного комплекса гистосовместимости класса I
-
Дефицит молекул главного комплекса гистосовместимости класса II
-
-
Преимущественные дефициты антител.
-
Агаммаглобулинемия с отсутствием В-клеток
-
Гипогаммаглобулинемия с нормальным или сниженным количеством В-клеток
-
Первичные В-клеточные дефекты. Синдромы, связанные с блоком переключения класса иммуноглобулинов
-
Селективный дефицит IgA
-
Другие первичные иммунодефициты, связанные с дефицитом изотипов или легких цепей иммуноглобулинов
-
Дефицит специфических антител с нормальной концентрацией иммуноглобулинов
-
Транзиторная гипогаммаглобулинемия детей раннего возраста
-
-
Синдромы иммунодефицитов с хорошо охарактеризованными клиническими признаками.
-
Синдром Луи-Бар
-
Синдром Неймеген
-
Синдром Вискотта-Олдрича
-
Синдром Ди Джорджи
-
Гипер-IgE-синдром
-
Хронический кожно-слизистый кандидоз
-
-
Генетические нарушения регуляции иммунитета.
-
Семейный гемофагоцитарный лимфогистиоцитоз
-
Иммунодефициты с гипопигментацией
-
Синдром Чедиака – Хигаси
-
Х-сцепленный лимфопролиферативный синдром
-
Аутоиммунный лимфопролиферативный синдром
-
Х-сцепленные иммунодисрегуляция, полиэндокринопатия, энтеропатия
-
-
Врожденные дефекты фагоцитов.
-
Тяжелые врожденные нейтропении
-
Циклическая нейтропения
-
Дефициты адгезии лейкоцитов
-
Хроническая гранулематозная болезнь
-
Дефицит глюкозо-6-фосфатдегидрогеназы нейтрофилов
-
Дефицит миелопероксидазы
-
-
Дефекты врожденного иммунитета: рецепторов и сигнальных компонентов.
-
Аутовоспалительные нарушения.
-
Дефициты комплемента.
-
Наследственный ангионевротический отек
-
“Настораживающие признаки”
-
Частый отит (6-8 раз в год и чаще)
-
Синусит (4-6 раз в год)
-
Пневмония (2 раза в год и чаще)
-
Абсцессы кожи и внутренних органов (особенно повторные)
-
Антибиотикотерапия для купирования инфекции с применением а/б в/м или per os в течение 2 мес и более, а также с использованием в/в а/б
-
Не менее двух перенесенных инфекций таких, как – менингит, сепсис, остеомиелит
-
Отставание ребенка в росте и массе тела (по возрастным нормам)
-
Персистирующая молочница или грибковые поражения кожи в возрасте старше 1 года
-
Указание на наличие в семье больных ПИД, ранние смерти детей от тяжелых инфекций
Общие особенности клинической картины
Ведущим в клинической картине ПИД является так называемый инфекционный синдром - повышенная восприимчивость к возбудителям инфекционных заболеваний в целом, необычно тяжёлое рекуррентное (рецидивирующее) их клиническое течение, наличие в этиологии заболевания атипичных возбудителей (часто оппортунистических). Тип возбудителя определяется характером иммунного дефекта. При дефектах антителообразования удаётся выявить устойчивую к антибактериальным препаратам флору - стафилококков, стрептококков, гемофильную палочку. При Т-клеточной иммунной недостаточности помимо бактерий выявляют вирусы (например, семейство герпесвирусов), грибки (Candida spp., Aspergillus и др.), а при фагоцитарных дефектах - стафилококки, грамотрицательные бактерии, грибки и т.д.
-
Врожденные дефекты фагоцитов.
Врожденная форма иммунной недостаточности с дефектами числа и функций фагоцитов, в частности нарушением подвижности, хемотаксиса, адгезии, эндоцитоза, киллинга, деградации фагоцитированных частиц, секреции цитокинов и др.
Тяжелые врожденные нейтропении
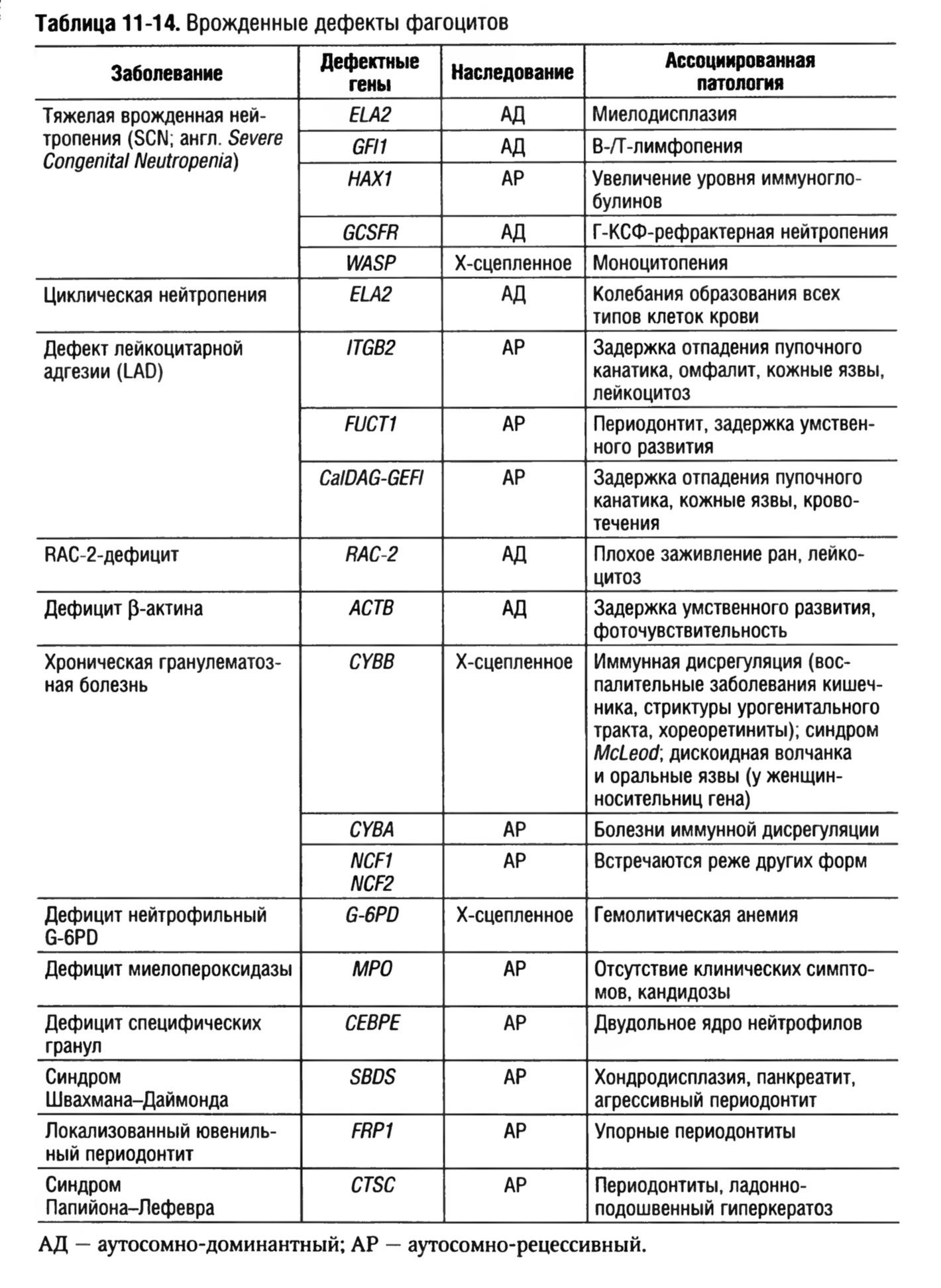
 Врожденные
нейтропении – это группа наследственных
патологий, которые передаются по
аутосомно-доминантному или
аутосомно-рецессивному типу и проявляются
уменьшением количества нейтрофилов в
периферической крови. Все заболевания,
входящие в эту группу, были описаны в
ХХ веке: синдром Костмана – в 1956 году,
семейная доброкачественная нейтропения
– в 1939 году, циклическая нейтропения –
в 1910 году, синдром «ленивых лейкоцитов»
– в 1964 году. Встречаются данные патологии
редко. Распространенность колеблется
от 1-2:100000 до 1 случая на 1 млн. младенцев.
Врожденными нейтропениями с одинаковой
частотой болеют как мальчики, так и
девочки. Прогноз зависит от формы
заболевания, при синдроме Костмана
летальность достигает 97-100%, в то время
как при семейной доброкачественной
нейтропении исход, как правило,
благоприятный.
Врожденные
нейтропении – это группа наследственных
патологий, которые передаются по
аутосомно-доминантному или
аутосомно-рецессивному типу и проявляются
уменьшением количества нейтрофилов в
периферической крови. Все заболевания,
входящие в эту группу, были описаны в
ХХ веке: синдром Костмана – в 1956 году,
семейная доброкачественная нейтропения
– в 1939 году, циклическая нейтропения –
в 1910 году, синдром «ленивых лейкоцитов»
– в 1964 году. Встречаются данные патологии
редко. Распространенность колеблется
от 1-2:100000 до 1 случая на 1 млн. младенцев.
Врожденными нейтропениями с одинаковой
частотой болеют как мальчики, так и
девочки. Прогноз зависит от формы
заболевания, при синдроме Костмана
летальность достигает 97-100%, в то время
как при семейной доброкачественной
нейтропении исход, как правило,
благоприятный.
Характеризуется ранними проявлениями бактериальных инфекций и персистирующей тяжелой нейтропенией.
Механизм развития
До конца не выяснен, мультигенные нарушения с различными формами наследования.