

Сосудистая хирургия часть 1
.pdfГлава 9. Атеросклероз: биологические и хирургические аспекты |
145 |
|
|
1.Эндотелиальные клетки выстилают просвет сосуда и служат для контроля сосудистого тонуса, продуцирования субстанций матрикса, таких как эластин, коллаген и протеогликаны. Слой эндотелиальных клеток служит для предотвращения тромбоза путем создания избирательного барьера между циркулирующей кровью и интерстициальной жидкостью.
2.Гладкомышечные клетки расположены глубже в артериальной стенке, составляя 40–50% объема медии в сосудах эластического типа и 80–85% в артериях мышечного типа. Гладкомышечные клетки поддерживают сосудистый тонус артериальной стенки и секретируют белки экстрацеллюлярного матрикса, такие как эластин, коллаген и гликозаминогликаны. Кроме того, обнаружено, что гладкомышечные клетки содержат рецепторы к липопротеинам и факторам роста и синтезируют простагландины для механической регуляции кровотока.
Эндотелиальные и гладкомышечные клетки in vivo обычно находятся в статичном состоянии. Эндотелий существует обязательно в виде монослоя благодаря контактному ингибированию. Гладкомышечные клетки, как было выяснено, имеют скорость обновления около 0,06% в сутки. Эти два типа клеток существуют совместно через сложную систему сигналов, влияющих на функцию друг друга. Например, эндотелиальные клетки секретируют продукт, который оказывает воздействие на функцию гладкомышечных клеток [19]. Вазодилатирующие субстанции, такие как простациклин, простагландин Е2 и эндотелий-зависимый расслабляющий фактор (EDRF), секретируются функционирующим эндотелием в ответ на локальное тромбогенное воздействие [20]. Это может объяснить то, что коронарные артерии, стенозированные до 40%, расширяются в ответ на изменения кровотока. Только после сужения просвета более 40% кровоток снижается (рис. 9.6) [21]. Утолщение сосудистой стенки зависит не только от пролиферации эндотелиальных клеток, но и накопления в гладкомышечных клетках и матриксе белков интимы. Было изучено несколько стимуляторов, и как будет обсуждено позднее, одним из наиболее сильных является тромбоцитарный фактор роста (PDGF).
Теории атеросклероза
Моноклональная гипотеза
Эта теория родилась на основе наблюдений, что индивидуальные клетки из атеросклеротических бляшек у женщин, гетерозиготных по Х-связанному гену глюкозо-6-фосфат дегидрогеназы (G-6PD), обычно проявляют только один G-6PD изотип (рис. 9.7) [22]. Это предполагает, что клетки из отдельной бляшки являются произ-
A
Б
Рис. 9.8. Распределение LDH изоэнзимов меди (А) и атеросклеротической бляшки (Б).
водными одной гладкомышечной клетки-предшественницы, и, хотя некоторые гладкомышечные клетки могут инфильтрировать интиму, основная масса клеток, обнаруженных в бляшке, вероятно, является результатом моноклональной пролиферации гладкомышечных клеток. Другое исследование подтвердило моноклональное поведение клеток в бляшках человека, используя LDH как маркер. Анализ изоэнзима LDH, раздельно проведенный на сосудах и бляшках человека, выявил сдвиг в распределении следов изоэнзима (рис. 9.8). Этот сдвиг отражает изменение типа гладкомышечных клеток, различия гладкомышечных клеток бляшки и интимы [23]. Такие данные дают дополнительную поддержку моноклональной гипотезы, однако не объясняют другие аспекты атеросклеротического процесса.
Гипотеза скопления интимальных клеток
Гипотеза основана на наблюдении, что у детей могут быть обнаружены небольшие скопления гладкомышечных клеток, где впоследствии развиваются атеросклеротические изменения. Остается неясным, как эти остатки развиваются, но они могут быть первоосновой пула клеток, подверженных атерогенезу. Эти скопления гладкомышечных клеток в сосудах детей широко выявляются в мире независимо от распространенности атеросклероза. Предполагается, что по существу развитие атеросклероза определяется внешними факторами, такими как повышенный уровень холестерина, курение и т. д. Группа патобиологического изучения атеросклероза молодого возраста (PDAY) исследовала этот феномен. Были собраны и изучены коронарные артерии, аорта и другие имеющие отношение ткани у лиц 15–34 лет. Исследователи сообщили, что жировые пятна в аорте были почти у всех исследуемых в возрасте от 15 лет и старше, фиброзные бляшки — у некоторых в возрасте 20 лет. Согласуясь с клиническими наблюдениями, было обнаружено, что коронарные артерии молодых мужчин имеют значительно большее количество выраженных бляшек по сравнению с женщинами [24].
Рис. 9.7. Энзимограмма образцов из: 1 — крови, 2 — нормальных тканей, 3-6 — атероматозных бляшек. В образцах из различных бляшек проявляются энзимы типа А или В, каждый из которых встречается в крови или нормальных тканях.
Гипотеза инкрустации
Эта гипотеза предполагает, что повторяющиеся циклы тромбоза и заживления служат источником прогрессирования бляшки. Так как известно, что тромбоз является поздним компонентом атеросклеротического поражения, данная теория не объясняет начало формирования бляшки [25].
Липидная гипотеза
Альтернативная гипотеза утверждает, что повышенный уровень липопротеидов низкой плотности (ЛПНП) приводит к патоло-
146 Раздел II. Основные сердечно-сосудистые проблемы
Эндотелий
Просвет
Макрофаги, |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
постоянно |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
находящиеся |
|
С |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
в интиме |
|
|
в |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
б |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
о |
о |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
д |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
н |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ы |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
е |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
р |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
а |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
д |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
и |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
к |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
а |
|
|
|
|
|
|
|
Накопление липопроте- |
|
Гладкомышечные |
Сво |
|
|
|
|
|
|
лы |
|
|
|
|
|
|||||
|
|
|
|
|
рад |
|
|
|
|
|
|
|
|
Окисленные липопротеины |
||||
|
|
|
бод |
|
|
|
|
|
|
|
|
|
|
|
|
|
в матриксе |
|
|
|
|
ные радикалы |
|
|
ы |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
л |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
а |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
к |
|
|
|
|
|
|
|
|
|
клетки |
|
С |
|
|
|
е |
и |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
ны |
|
|
|
|
|
окисления |
|
|
||||
|
|
|
|
|
од |
|
|
|
|
|
|
|
Начало |
низкой плотности |
||||
|
|
|
воб |
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
протеинов |
в |
липо- |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
виях |
их |
|
усло- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ного |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
повышен- |
|
|||
|
|
|
|
|
|
|
|
|
Интима |
содержания |
|
|||||||
Внутренняя
эластическая
мембрана
Медиа
Хемоаттрактанты Моноциты моноцитов
Повреждение клетки
Проникновение повышенных концентраций липопротеинов низкой и очень низкой плотности
Гладкомышечные |
клетки |
|
Окисленные липопротеины низкой плотности
Рецепторно
опосредованные
каналы
Образование агрегатов и фагоцитоз
Повреждение |
Окисленные липопротеины |
|
низкой плотности |
||
|
||
|
клетки |
Моноциты, производ- |
|
ные макрогфагов |
|
(пенистые клетки) |
- |
|
|
гладко |
|
клеток |
|
Пролиферация |
|
мышечных |
|
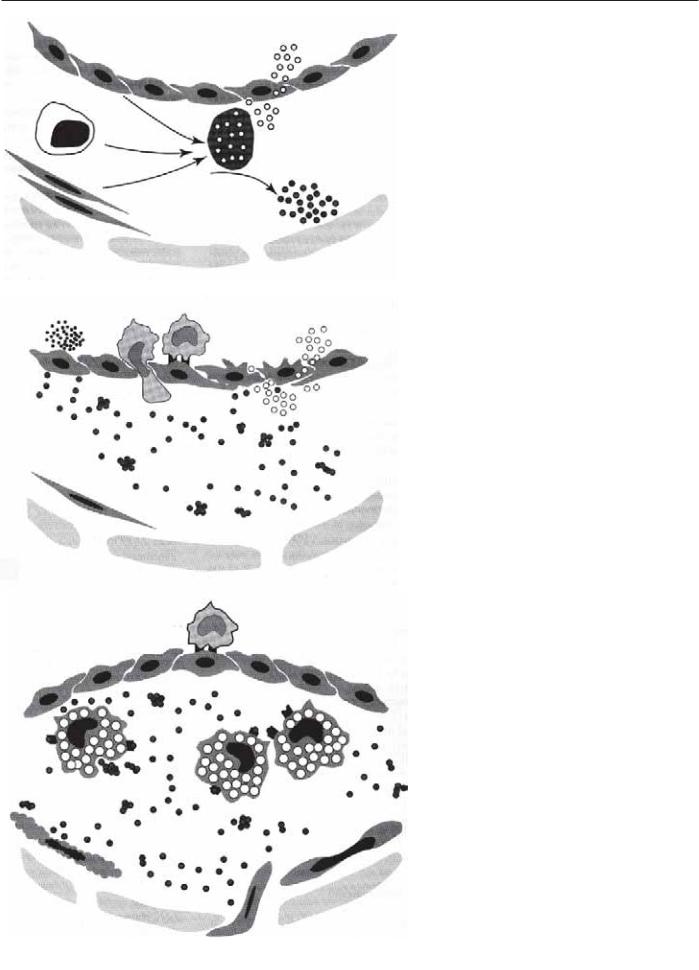
Рис. 9.9. Схематическая последовательность развития атеросклероза под воздействием окисленных липопротеинов.
Глава 9. Атеросклероз: биологические и хирургические аспекты |
147 |
||
|
|
||
гическому накоплению липидов в гладкомышечных клетках и |
2) повышенную продукцию экстрацеллюлярного матрикса; |
||
макрофагах при проникновении через сосудистую стенку [26, |
3) агрегацию липидов. |
|
|
27]. Так как ЛПНП окислены, происходит повреждение эндо- |
Эти процессы приходят в движение, когда эндотелий сосуда |
||
телиальных клеток, и атерогенные процессы, о которых гово- |
подвергается какому-либо повреждению. Постоянное поврежде- |
||
рилось ранее, ведут к формированию бляшки. Установлено, что |
ние эндотелия вызывает очаговый хронический воспалитель- |
||
окисленные липопротеины являются причиной повреждения |
ный ответ, результатом которого является развитие атероскле- |
||
клеток независимо от способа окисления [28–31]. Окисление ли- |
ротической бляшки (рис. 9.11). В самом деле, все упомянутые |
||
попротеинов ведет к образованию нескольких токсических про- |
теории пытаются объяснить развитие атеросклероза, но, в лучшем |
||
дуктов, включая 7-b-гидропероксихолестерол, 7-кетохоле-сте- |
случае, помогают объяснить только отдельные аспекты очень слож- |
||
рол, лизоферменты, окисленные жирные кислоты и эпоксистерол |
ного процесса [39–41]. |
|
|
[32, 33]. Точный механизм гибели клетки все еще неизвестен. На |
|
|
|
рис. 9.9 приведена одна из теорий развития атеросклероза, вы- |
Морфология и гемодинамика |
|
|
званного окисленными ЛПНП [34]. Липопротеины очень низ- |
|
||
кой плотности (ЛПОНП) и ЛПНП накапливаются в интиме. По- |
|
|
|
вышенные уровни липопротеинов и связь с элементами |
Следует повторить, что артериальные сосуды подвергаются боль- |
||
соединительной ткани увеличивают время пребывания липопро- |
шим гемодинамическим нагрузкам, которые оказывают сильное |
||
теинов в интиме, тем самым повышая вероятность подвергнуть- |
ударное воздействие на внутреннюю эндотелиальную выстилку. |
||
ся окислению [35,36]. После окисления модифицированные ли- |
Монослой из эндотелиальных клеток активно участвует в слож- |
||
попротеины стимулируют проникновение моноцитов и |
ном взаимодействии между кровью, находящейся в просвете, и са- |
||
лимфоцитов в интиму. Окисленные ЛПНП также способствуют |
мой сосудистой стенкой. Действительно, биологический ответ эн- |
||
миграции и пролиферации гладкомышечных клеток, что вносит |
дотелия на гемодинамические нагрузки является основным |
||
вклад в происхождение атеросклеротической бляшки. Недавно |
вопросом развития атеросклероза. Артериальный сосуд подвер- |
||
выяснены доказательства в пользу этой гипотезы. Более точно |
гается двум первичным гемодинамическим нагрузкам: напряже- |
||
показано, что липиды, обнаруженные в бляшке, поступают пря- |
нию сдвига и циклической деформации растяжения (рис. 9.12). |
||
мо из кровотока и есть существенные доказательства связи ги- |
При движении крови вдоль эндотелия возникает направленное по |
||
перхолестеринемии и повышенной склонности развития ате- |
касательной усилие торможения, называемое напряжением сдви- |
||
росклеротических изменений [37, 38]. Повышенный уровень |
га [42, 43]. Величина напряжения сдвига прямо пропорциональ- |
||
ЛПНП плазмы ведет к повышению уровня ЛПНП в интерсти- |
на вязкости крови и обратно пропорциональна радиусу сосуда в |
||
циальной жидкости, где происходит связь с протеогликанами. |
третьей степени. Исследования показали, что высокое напряже- |
||
Накопление ЛПНП увеличивает сродство липопротеинов к окис- |
ние сдвига обратно пропорционально распространенности ранних |
||
лению. Последнее, как было выяснено, является причиной по- |
интимальных поражений. Те зоны, которые подвержены повышен- |
||
вышенной продукции PDGF гладкомышечными клетками, по- |
ному напряжению сдвига, оказались более защищенными и име- |
||
вышенной гибели гладкомышечных клеток, замедленного |
ли меньшие интимальные изменения в сравнении с зонами с низ- |
||
заживления эндотелия, а также пролиферации моноцитов по- |
ким или колеблющимся уровнем напряжения сдвига (рис. 9.13) |
||
средством повышенной секреции моноцитарного хемоаттрактан- |
[44–46]. Эти данные привели к многочисленным исследованиям, |
||
та протеина-1 (МСР-1) эндотелиальными клетками [27]. |
пытающимся охарактеризовать влияние гемодинамики на сосу- |
||
|
дистую биологию. Результаты этих исследований продемонстри- |
||
Гипотеза реакции на повреждение |
ровали несколько возможных типов ответной реакции на напря- |
||
жение сдвига (табл. 9.3). Например, было обнаружено, что |
|||
|
|||
Процесс атеросклероза является хроническим, действующим ис- |
эндотелиальные клетки изменяют ориентацию по кровотоку под |
||
подволь и проявляющимся после нескольких десятилетий жизни. |
воздействием напряжения сдвига. Кроме того, реорганизация эн- |
||
Начало процесса объясняют несколько теорий. Одна из них воз- |
дотелиального F-актина в цитоскелете позволяет возникать мор- |
||
никла из наблюдения, при котором обнаружено, что дисфунк- |
фологическим изменениям под влиянием напряжения сдвига (рис. |
||
ция эндотелиальных клеток ведет к атеросклерозу. В результате |
9.14) [47–50]. Как показано на рисунке, выступающие пучки ми- |
||
дисфункции эндотелиальных клеток возникают повышенная со- |
крофиламентов актина локализуются и упорядочиваются в зонах |
||
судистая проницаемость, повышенная адгезия лейкоцитов, функ- |
высокого напряжения сдвига. В зонах с низким уровнем напряже- |
||
циональный дисбаланс про- и антитромботических факторов, мо- |
ния сдвига и неламинарным характером кровотока пучки актино- |
||
дуляторов роста и вазоактивных субстанций (рис. 9.10). Эта |
вых монофиламентов остаются плотными и неупорядоченными. |
||
начальная дисфункция эндотелиальных клеток также иницииру- |
Было показано, что напряжение сдвига ингибирует миграцию и |
||
ет прогрессирование атеросклероза. Накапливающиеся в зоне по- |
пролиферацию эндотелиальных клеток [51]. Наконец, напряже- |
||
вреждения лейкоциты высвобождают больше факторов роста, те, |
ние сдвига воздействует на биологическое функционирование эн- |
||
в свою очередь, индуцируют миграцию гладкомышечных клеток |
дотелиальных клеток, доказывая их роль в защите сосудов от ате- |
||
в интиму. Гипотеза реакции на повреждение также утверждает, что |
росклероза. Напряжение сдвига увеличивает секрецию |
||
тромбоциты, присутствующие в зоне поврежденного эндотелия, |
простациклина, который действует как мощный вазодилататор |
||
секретируют мощный митогенный фактор, тем самым стимули- |
и ингибитор агрегации тромбоцитов [52, 53]. Аналогично секре- |
||
руя пролиферацию гладкомышечных клеток. Гипотеза объединя- |
ция активатора тканевого плазминогена, сильного тромболитика, |
||
ет в себе три важных процесса, вовлеченных в атерогенез: |
и оксида азота, сильного медиатора вазомоторного тонуса и глад- |
||
|
комышечной пролиферации, усиливается с повышением уровня |
||
1) происходящие в интиме очаговые процессы миграции, проли- |
напряжения сдвига [54, 55]. Эти факты предполагают наличие ме- |
||
ферации и аккумуляции различных клеток, таких как мак- |
ханизма, с помощью которого можно объяснить повышенный ате- |
||
рофаги и гладкомышечные клетки; |
рогенез в зонах с низким напряжением сдвига. |
|
|
148 Раздел II. Основные сердечно-сосудистые проблемы
|
|
Окисленные липиды/ |
|
Активация |
|
|
|
|
|
|
|
свободные радикалы |
|
|
|
Вирусная инфекция |
|
Напряжение |
Цитокинез |
|
Гипоксия |
сдвига |
||
Тромбин |
|
|
||
|
|
|
||
|
Гомоцистеин |
|
|
|
Эндотелиальные
клетки
|
|
Ответные реакции |
|
Изменение |
Вазоактивные |
Адгезия лейкоцитов |
проницаемости |
субстанции |
|
|
Прокоагулянтная |
Факторы роста/ |
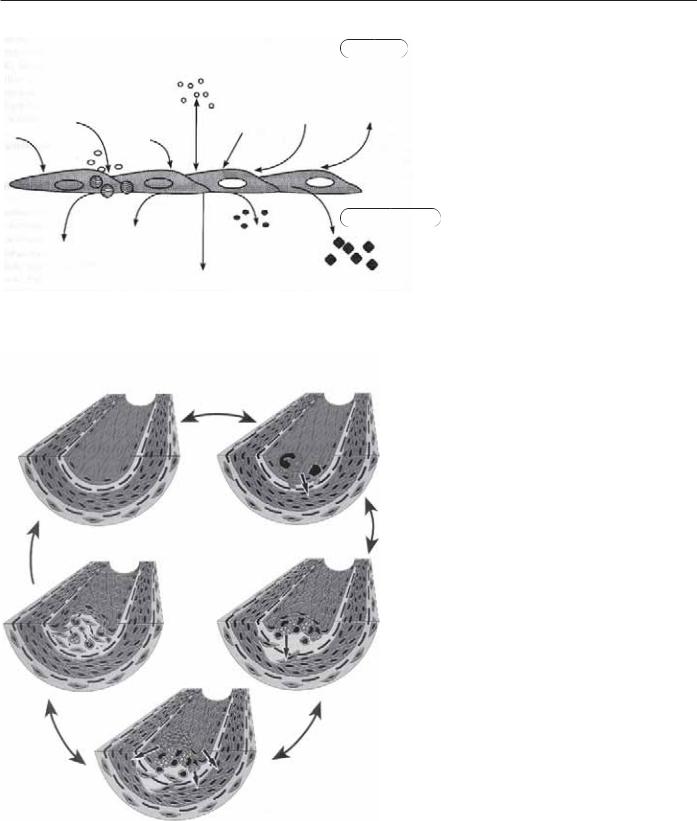
Рис. 9.10. Дисфункция эндотелия при повре- |
|
активность |
ждении. |
||
хемоаттрактанты |
«ПОВРЕЖДЕНИЕ» Окисленные липопротеины низкой плотности (механическое, гомоцистеиновое, иммунологическое, токсическое, вирусное и т. п.)
Рис. 9.11. Гипотеза реакции на повреждение. Известно, что каждая стадия потенциально обратима при устранении повреждающего агента.
К циклической деформации относятся повторяющиеся цир- |
деформацию (табл. 9.3). Происходящие морфологические измене- |
кулярные пульсовые растяжения сосудистой стенки. Вместе с на- |
ния вторичны по отношению к реорганизации актина в цитоске- |
пряжением сдвига эндотелиальные клетки специфическим обра- |
лете и приводят к организованному расположению клеток, перпен- |
зом реагируют на циклическую деформацию растяжения. Было |
дикулярно вектору силы [56, 57]. Также обнаружено, что |
показано, что культура эндотелиальных клеток пролиферирует и |
циклическая деформация стимулирует некоторые макромолеку- |
проявляет морфологические изменения в ответ на циклическую |
лы. Под воздействием напряжения сдвига и циклической дефор- |

Глава 9. Атеросклероз: биологические и хирургические аспекты |
149 |
|
|
2
1
1
Пульс
Зона высокого напряжения сдвига
2
Зона низкого напряжения сдвига
Поток Поток
Рис. 9.12. Схема гемодинамических сил, возникающих во вре- |
Поперечный срез |
мя систолы. Вектор силы напряжения сдвига параллелен кро- |
каротидного синуса |
|
|
вотоку и однонаправлен. В противоположность ему, вектор си- |
|
лы циклической деформации растяжения многоплановый и |
|
разнонаправленный. (1) Растягивающее усилие. (2) Напряжение |
|
сдвига. |
Рис. 9.13. Графическое распределение кровотока в области |
|
каротидной бифуркации. |
A |
|
Б |
|
|
|
В |
|
Г |
|
|
|
Таблица 9.3. Влияние гемодинамики на функцию клеток
Рис. 9.14. Морфологические изменения микрофиламентов актина в аорте кролика под воздействием напряжения сдвига. (А) Грудная аорта. (Б) Низкое напряжение сдвига. (В) Высокое напряжение сдвига. (Г) Электронная микрофотография эндотелиальных клеток, подвергающихся высокому напряжению сдвига. Обращает внимание выступающий пучок микрофиламентов актина.
Клеточная функция |
Воздействие напряжения сдвига |
Воздействие циклического растяжения |
|
|
|
Пролиферация |
Ингибирование пролиферации эндотелиальных |
Стимуляция пролиферации гладкомышечных |
|
и гладкомышечных клеток |
клеток |
Ориентация |
Эндотелиальные клетки вытягиваются и линейно |
Эндотелиальные клетки ориентируются |
|
ориентируются вдоль кровотока |
перпендикулярно к направлению вектора усилия |
Секреция |
Стимуляция секреции окиси азота, PGI2, tPA |
Стимуляция секреции окиси азота, PGI2, tPA |
Сигнальная трансдукция |
Активация путей DAG/IP3, интегрирования |
Активация путей DAG/IP3, МАР-киназ и TGF-β |
|
и МАР-киназ |
|
|
|
|

150 Раздел II. Основные сердечно-сосудистые проблемы
мации эндотелиальные клетки продуцируют в повышенном количестве простациклин и tPA. В дополнение к этому, эндотелиальный синтез оксида азота и, следовательно, его уровень также повышаются [58–60]. Более того, циклическая деформация стимулирует проявления клеточной адгезии таких молекул, как ICAM-1 [61]. Исследования показали, что система вторичных месенджеров, таких как аденилатциклазный-сАМР и дициклоглице- роловый-IP3 путь, активизируются циклической деформацией [62]. Хотя все еще неясно, как эти данные могут подвести к пониманию механизмов ответа эндотелиальных клеток, служащих промежуточным звеном в развитии атеросклероза.
В поддержку решающей роли гемодинамических сил следует отметить, что атеросклеротические поражения не возникают случайно по всей сосудистой системе [63]. Майкл ДеБеки и соавторы [64] выделили пять бассейнов распространения атеросклеротических поражений. Они отметили, что коронарные артерии, ветви дуги аорты, брюшная аорта, крупные висцеральные артерии и артерии нижних конечностей особенно подвержены атеросклерозу. Клинические проявления заболевания в большинстве случаев связаны с локализацией атеросклеротических бляшек в этих сосудистых участках (рис. 9.15) [65].
1.Категория 1 включает коронарные артерии, которые содержат много ответвлений, подверженных скручиванию при каждом сокращении сердца. Атеросклеротические поражения обычно возникают в местах бифуркаций крупных артерий, например в месте деления левой коронарной артерии на левую переднюю нисходящую и левую огибающую артерии.
Проксимальное
Среднепроксимальное
Дистальное
Рис. 9.15. Преимущественная локализация атеросклеротических поражений.
2.Категория 2 включает ветви дуги аорты. Сонные артерии особенно подвержены атеросклерозу.
3.Категория 3 состоит из висцеральных ветвей брюшной аорты. Чревный ствол, верхняя и нижняя брыжеечные артерии и почечные артерии считаются наиболее восприимчивыми.
4.Категория 4 включает дистальный отдел брюшной аорты и подвздошные артерии. Большинство пациентов с атеросклеротической патологией попадает в эту категорию. Кроме того, пациенты с симптоматическим поражением терминальной аорты и артерий нижних конечностей имеют наиболее высокую вероятность атеросклеротического поражения других локализаций [64].
5.Категория 5 включает пациентов, имеющих одновременно поражение в двух и более вышеупомянутых бассейнах.
Известно, что поверхностная бедренная артерия (ПБА) в приводящем канале имеет особую склонность к атеросклеротическому поражению, хотя никаких ответвлений на этом участке нет. У пациента могут появиться симптомы при стенозе этой артерии вследствие недостаточности компенсаторных возможностей [66]. Как упоминалось выше, требуется сужение просвета сосуда не менее 40% по площади, прежде чем появятся симптомы, поскольку компенсаторная дилатация пытается поддержать кровоток. Вследствие анатомического сужения ПБА не способна к компенсации, и у пациентов могут появиться симптомы и при меньшем поражении.
Кровоток в артериях человека обычно ламинарного характера. На прямом отрезке, не имеющем ответвлений, кровоток ламинарный и параболический. Скорость максимальная в центре просвета и наименьшая в зоне соприкосновения крови с эндотелием за счет силы трения. Бифуркация и другие геометрические изменения влияют на локальные характеристики кровотока, вызывая турбуленцию. При турбуленции профиль кровотока беспорядочный и хаотичный и зависит от вязкости крови, средней скорости и диаметра сосуда [67].
В области бифуркаций сосудистого русла имеется повышение напряжения сдвига и турбулентности [46]. Вектор скорости в этих зонах становится нелинейным. Может быть, именно этими изменениями гемодинамических факторов, возникающими в области бифуркаций, можно объяснить топографическую избирательность атеросклероза. Исследования продемонстрировали, что митотическое деление эндотелиальных клеток на 50% происходит чаще в зонах с турбулентным кровотоком по сравнению с участками ламинарного кровотока [68]. Более того, гиперхолестеринемия может снижать устойчивость эндотелиальных клеток, делая их более чувствительными к гемодинамическим воздействиям, особенно в местах ответвлений [69].
Стадии атеросклероза
Начальная стадия
Как только процесс атеросклероза инициируется, возникает цепь последовательных событий, результатом которых является образование жировых пятен, рассматриваемых как самые ранние проявления атеросклеротического поражения (рис. 9.16). Повышенная сосудистая проницаемость ведет к накоплению липидов и развитию атерофиброзной бляшки (рис. 9.17) [70]. После экспериментов с кроликами было предположено, что пролонгированное время нахождения крови в зоне сниженного напряжения сдвига может способствовать увеличению миграции липопротеинов в
Глава 9. Атеросклероз: биологические и хирургические аспекты |
151 |
|
|
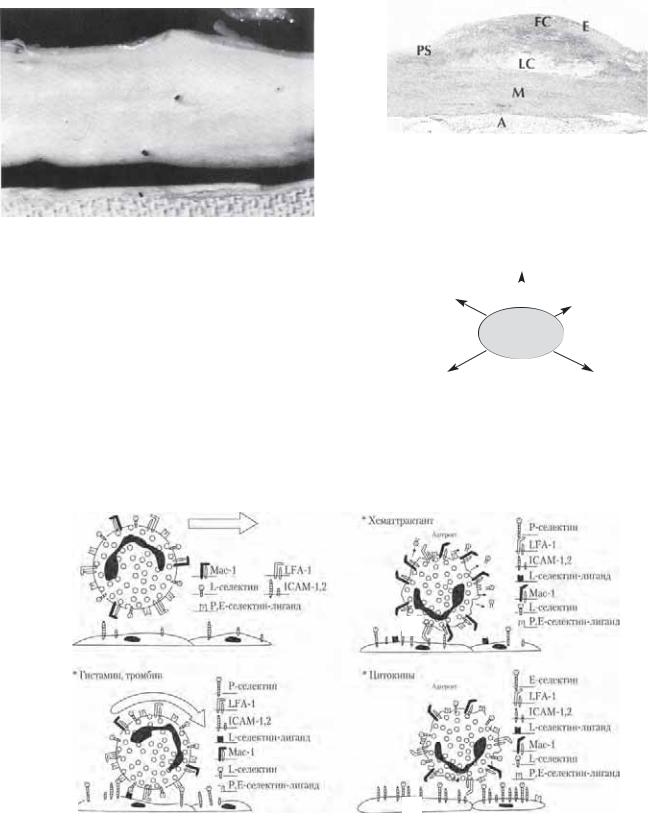
Рис. 9.16. Жировые пятна в грудной аорте.
интиму [37, 38]. Эти липопротеины, находящиеся в кровотоке, связываются с молекулами протеогликанов экстрацеллюлярного матрикса артерий и окисляются. Оба компонента липопротеинов модифицируются в продукты, активизирующие атерогенез. Липиды окисляются в гидропероксиды, оксистеролы и другие соединения [33]. Подобным образом модифицируются белки и распадаются на компоненты, которые до конца не охарактеризованы. Предполагается, что процессы окисления осуществляются системами НАД, НАДФ и липооксигеназами, обнаруженными в зоне формирования атеромы [71, 72]. Другие факторы, такие как гиперхолестеролемия, гомоцистеинемия и табакокурение, также способствуют усилению окисления липопротеинов [73–76]. Изве-
Свободно скользящий гранулоцит
A
|
|
|
|
|
|
йся г |
ра |
|
|
|
|
|
|
|
|
|
|
|
и |
н |
|
|
|
|
|
||
|
|
|
|
щ |
|
|
у |
|
|
|
|
||
|
|
|
ю |
|
|
|
|
|
|
|
|
||
|
|
а |
|
|
|
|
|
л |
|
|
|
||
|
щ |
|
|
|
|
|
|
|
о |
|
|
||
|
а |
|
|
|
|
|
|
|
|
|
ц |
|
|
р |
|
|
|
|
|
|
|
|
|
|
и |
||
В |
|
|
|
|
|
|
|
|
|
|
|
|
т |
Б
Рис. 9.17. Фиброзножировая бляшка в аорте человека. А –ад- вентиция, М — медия, LC — липидное ядро, PS — края бляшки, Е — эндотелий, FC — фиброзная покрышка.
|
|
Агрегация |
|
||
|
|
|
|
|
|
Гликозилирование |
|
|
|
|
Участие в образовании |
|
|
|
|
|
иммунных комплексов |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Липо- |
|||
|
|
протеины низ- |
|||
|
|
кой плотности |
|||
|
|
(ЛПНП) |
|||
|
|
|
|
|
|
Окисление |
|
|
|
|
Участие в образовании |
|
|
|
|
протеогликановых комплексов |
|
|
|
||||
Рис. 9.18. Возможные пути метаболизма ЛПНП, ведущие к инициации атеросклероза.
В
Г
Рис. 9.19. Последовательность этапов адгезии гранулоцитов. (А) В отсутствие воспаления гранулоциты не взаимодействуют с эндотелием. (Б) При воспалении эндотелиальные клетки быстро выделяют селектины и гранулоциты начинают вращаться. (В) Под воздействием хемоаттрактантов гранулоциты приобретают более адгезивное строение и связываются с белками ICAM. (Г) Гранулоциты способны к связыванию.

152 Раздел II. Основные сердечно-сосудистые проблемы
стно, что у пациентов с сахарным диабетом липопротеины также подвергаются неферментативному гликозилированию, что приводит к образованию продукта, способствующего развитию атеросклероза (рис. 9.18) [77, 78].
После повреждения эндотелия усиливается клеточная адгезия таких молекул, как VCAM-1, ICAM-1 и Е-селектина. Молекулы клеточной адгезии состоят из двух основных групп: суперсемейства иммуноглобулинов (VCAM, ICAM) и мембран-связанных гликопротеинов (селектинов). Было выяснено, что VCAM-1 взаимодействует с поздно образующимся антигеном-4 (VLA-4), специфическим интегрином, обнаруженным на лейкоцитах, моноцитах и Т-лимфо- цитах, обычно присутствующим в атеросклеротических бляшках. ICAM-1, другой тип адгезивных молекул, вероятно, играет роль в образовании атеромы, выступая в роли рецептора для LAF-1 и Mac-1 интегринов, обнаруживаемых на самых различных типах лейкоцитов. Селектин эндотелиальных клеток, или Е-селектин, является мембрано-связанным гликопротеином, который также, вероятно, играет заметную роль в сцеплении лейкоцитов. Е-селектин служит связующим звеном в сцеплении между нейтрофилами и эндотелиальными клетками. Этот тип адгезивных молекул вместе с другими селектинами, такими как Р-селектин (тромбоцитарный) или L-се- лектин (лейкоцитарный), содействуют скачкообразному продвижению лейкоцитов к эндотелию (рис. 9.19) [79].
Временное проявление каждой из этих адгезивных молекул заметно отличается и дает возможность проникнуть в суть понимания их специфических ролей в адгезии лейкоцитов (рис. 9.20) [80]. Е-селектин появляется через 1–2 ч после активации цитокинов и достигает пика своего проявления в период от 4 до 6 ч. VCAM-1 появляется через 4–6 ч после активации и достигает пиковой активности через 12–18 ч. Появление ICAM-1 занимает промежуточное положение с пиковой активностью через 4–6 ч. Следует отметить, что тогда как Е-селектин подвергается быстрой деградации через 6 ч даже в присутствии цитокинов, проявление ICAM-1 сохраняется весь период существования цитокинов, а проявление VCAM-1 убывает в течение нескольких дней. Эти временные различия пиковых проявлений и деградации могут означать прогрессирование адгезии эндотелиальных клеток. Е-селектин первым начинает процесс вращения лейкоцитов, уступая дорогу ICAM-1 и, наконец, VCAM-1, наращивая изменения в сторону более постоянной адгезии лейкоцитов [79]. Эти усиливающиеся взаимодействия между эндотелиальными клетками и лейкоцитами могут воз-
TNF-α |
Е-селектин |
VCAM-1 |
VCAM-1 |
|
|
|
|
50 Ед/мл |
|
|
|
0 ч |
|
|
|
6 ч
24 ч
Рис. 9.20. Временное проявление молекул клеточной адгезии к эндотелиальным клеткам пупочной вены человека (HUVEC), стимулированной фактором TNF-α.
вещать начало инфильтрации лейкоцитов в интиму и начало атеросклеротического процесса.
Внедавних исследованиях сообщается, что компоненты окисленных липопротеинов, такие как лизофосфатидилхолин, могут усиливать адгезивные свойства этих молекул [81]. Кроме того, на участках, где нарушаются ламинарные перемещения кровотока, проявления VCAM усиливаются [82].
Эти молекулы клеточной адгезии усиливают слипание в области повреждения сосуда и агрегацию моноцитов и Т-лимфоцитов. Лимфоциты мигрируют в сосудистую стенку и накапливаются в интиме. Окисленные ЛПНП и хемоаттрактант моноцитов проте- ин-1 (МСР-1) (оба являются продуктами эндотелиальных клеток
вокисленном состоянии) действуют как клеточный хемоаттрактант для интерлейкинов и других цитокинов [83, 84]. Это состояние воспаления приводит к повышению уровня альфа-фактора некроза опухолей (TNF-α) и интерлейкина-1 (IL-1), которые являются известными усилителями слипания лейкоцитов и ускоряют атеросклеротический процесс. Хотя механизм действия цитокинов уточняется, некоторые связи уже объяснены. Было показано, что TNF-α и IL-1 усиливают проявления ICAM-1 и VCAM-1. Эти цитокины вместе с факторами роста секретируются макрофагами и также вовлекаются в процессы миграции и пролиферации гладкомышечных клеток (рис. 9.21) [41, 85].
Однако данный процесс не остается бесконтрольным. Есть доказательства, что окись азота, высвобождаемая из эндотелия, служит ограничителем проявлений VCAM-1 даже при низком уровне [86]. L-аргинин метаболизируется в окись азота и побочный продукут L-цитруллин. Происходит это посредством индуцируемого синтеза эндотелиальной синтазы окиси азота (eNOS), которая является мембран-связанной кальций-кальмодулиново-за- висимым ферментом [87–89]. eNOS может регулироваться локальной гормональной активностью через брадикинин, АТФ и гистамин. Кроме того, по некоторым данным, гемодинамические факторы, такие как напряжение сдвига и циклическая деформация, увеличивают количество eNOS [59]. Следовательно, в зонах поврежденного эпителия окись азота может служить связующим звеном между вазомоторным контролем и пролиферацией гладкомышечных клеток.
Винтиме моноциты дифференцируются в макрофаги и накапливают липиды, превращаясь в пенистые клетки по мере возрастания содержания липидов. Эти клеточные очистители эндоцитоза модифицируют липопротеины посредством не-ЛПНП-специфического рецепторного пути и пытаются удалить их из интимы. Однако вследствие дисбаланса между накоплением липидов и удалением пенистых клеток происходит сетевое накопление в интиме [18]. Процесс «поглощения» липопротеинов макрофагами ведет к высвобождению цитокинов, которые стимулируют миграцию и пролиферацию гладкомышечных клеток (рис. 9.22) [41].
Жировые пятна предшествуют развитию более выраженной атероматозной бляшки. Микроскопически, жировые пятна в большом количестве содержат моноциты и Т-лимфоциты. Их можно увидеть невооруженным глазом в виде полосок или пятен на сосудистой стенке [70]. Однако не все жировые пятна достигают стадии проблемного поражения. В некоторых исследованиях выяснено, что жировые пятна существуют даже в сосудах плода человека. Они регрессируют в ранний период жизни и затем появляются вновь в детском возрасте [90]. Интересно, что женщины склонны к более раннему появлению и заметно большему количеству жировых пятен в аорте, чем мужчины. Этот факт является контрастом развития более выраженных поражений у мужчин, но

|
|
|
|
|
Глава 9. Атеросклероз: биологические и хирургические аспекты |
153 |
||
|
|
|
|
|
|
|
|
|
|
Нейтрофил |
|
|
|
|
|
|
|
|
LFA-1 |
|
МАС-1 |
|
|
|
|
|
|
|
Сиализированный |
|
|
|
|
||
|
|
|
|
|
|
|
||
|
|
|
углеводный |
|
|
|
|
|
|
PSGL-1 |
|
лиганд |
|
|
|
|
|
|
|
|
L-селектин |
|
|
|
МАС-1 |
|
Р-селектин |
Е- |
|
МАС-1 |
|
LFA-1 |
|
||
|
LFA-1 |
|
|
|||||
|
|
селектин |
|
|
|
|
||
|
|
ICAM-1 |
ICAM-1 ICAM-1 |
ICAM-1 |
|
|||
|
|
|
|
|
||||
PECAM-1 |
Рис. 9.21. Схематическое изображение ад- |
|
гезии и трансмиграции нейтрофилов. |
||
|
||
Эндотелий |
|
IFN-γ |
|
|
GM-CSF |
Апоптоз |
|
TNF-α |
||
|
||
Т-лимфоцит |
GM-CSF |
|
Модификация |
||
M-CSF |
||
липопротеинов |
|
|
Пролиферация |
IL-2 |
|
Ag |
||
|
Поглощение и очистка посторонних примесей
Индуцируемые источники молекул-регуля- Макрофаг торов роста
Агонисты |
Антагонисты |
Хемоат- |
роста |
роста |
трактанты |
GM-CSF |
IFN-γ |
GM-CSF |
M-CSF |
IL-1 |
M-CSF |
HB-EGF |
TGF-β |
VEGF |
IGF-I |
|
bFGF |
VEGF |
|
MCP-1 |
bFGF |
|
TGF-β |
IL-1 |
|
PDGF |
TNF-α |
|
ox LDL |
TGF-α |
|
|
TGF-β |
|
|
PDGF |
|
|
M-CSF/GM-CSF |
|
M-CSF/GM-CSF |
Ag |
|
HB-EGF |
Ag |
IL-1 |
|
|
VEGF |
TNF-α |
|
|
|
|
|
|
||
IL-1 |
|
MCP-1 |
|
|
TNF-α |
|
ox LDL |
|
Рис. 9.22. Потенциальная роль макрофагов в |
MCP-1 |
|
|
|
|
ox LDL Гладкомышечные клетки |
Эндотелиальные клетки |
атерогенезе. |
||
в более поздний период жизни [91, 92]. Эти противоречия делают взаимосвязь между жировыми пятнами и прогрессированием атеросклероза затруднительной и запутанной. Однако возможно, что события, которые следуют за развитием жировых пятен, вовлекая гладкомышечные клетки, предвещают развитие клинически значимого атероматозного поражения.
урокиназного активатора плазминогена (uPA), tPA и ингибитора активатора плазминогена 1 (PAI-1) — важных факторов сосудистой репарации [102]. Обнаружены также другие вещества с митогеной активностью — IL-1, тромбин и TNF-α, действие которых осуществляется непрямым путем через стимуляцию активности PDGF [103–105].
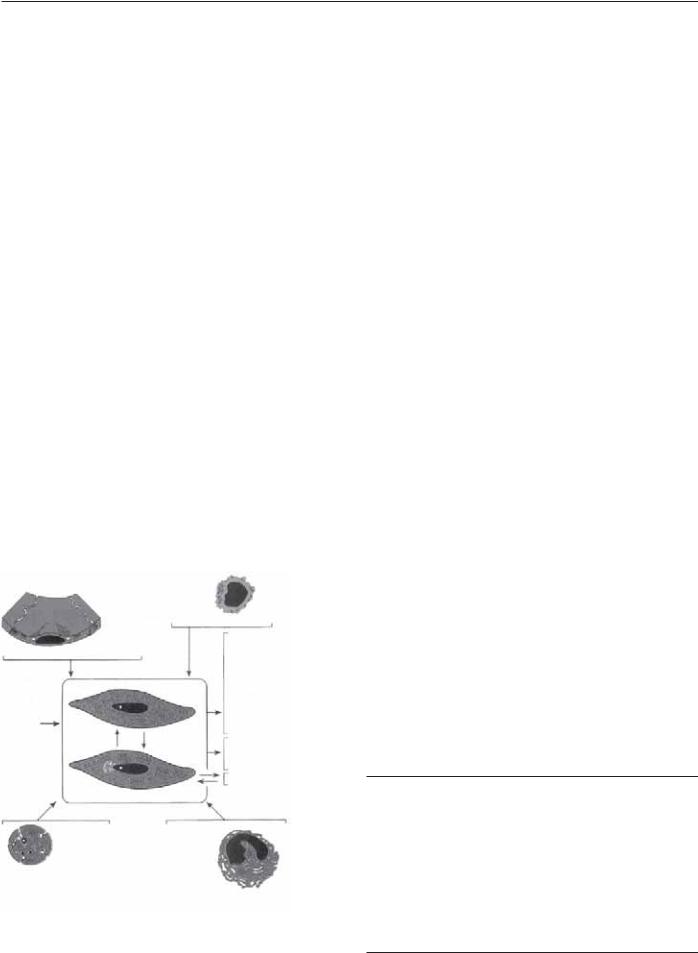
Прогрессия
В то время как дисфунция эндотелиальных клеток является основой формирования атеросклеротического поражения, пролиферация гладкомышечных клеток является важным фактором развития (рис. 9.23). Хемоаттрактанты, такие как PDGF, индуцируют миграцию гладкомышечных клеток из медии в интиму [94–96]. После миграции пролиферация и рост гладкомышечных клеток стимулируются факторами роста, которые включают фактор роста фибробластов (FGF), гепарин-связанный эпидермальный фактор роста (HB-EGF), PDGF и TGF-β [97–101]. Фактор роста сосудистых эндотелиальных клеток (VEGF) является еще одной субстанцией, которая, как обнаружено, обладает сильным митогенным воздействием на эндотелиальные клетки. VEGF продуцируется эндотелиальными клетками и макрофагами и стимулирует эндотелиальные клетки к продукции коллагеназы,
IL-1
TNF-α
TGF-β
|
Эндотелиальные клетки |
PDGF |
PDGF-AA |
Активация |
PDGF |
|
|
макрофагов |
|
Гладкомышечные клетки
Рис. 9.23. Пролиферация гладкомышечных клеток.
154Раздел II. Основные сердечно-сосудистые проблемы
Вдополнение к факторам роста и цитокинам, факторы коагувключает коллаген 1 и 3 типов, эластин и протеогликаны [41].
ляции также вносят свой вклад в развитие атеросклеротического поражения. В зонах повреждения эндотелия формируются микротромбы, богатые тромбоцитами. Активированные тромбоциты, содержащиеся в области повреждения, высвобождают несколько факторов, поддерживающих ответную фиброзную реакцию [92, 106, 107]. По мере созревания бляшки, из vasa vasorum развиваются и распространяются мелкие сосуды. По этой системе микроциркуляции происходит доставка веществ, поддерживающих развитие атеромы. Кроме того, могут происходить локальные кровоизлияния с высвобождением тромбина в области бляшки. Помимо свертывания крови, тромбин модулирует активность гладкомышечных клеток через активность PDGF-фактора, таким образом поддерживая процесс накопления в интимальных клетках в ответ на повреждение [108].
Гладкомышечные клетки, попавшие в интиму, медленно пролиферируют в течение десятилетий с периодами усиленного клеточного деления, вызванными периодическими разрывами бляшки [109]. Однако эта пролиферация регулируется локальными цитостатическими медиаторами, такими как TGF-β и IFN-γ, которые ингибируют деление гладкомышечных клеток [110, 111]. Также имеются данные в поддержку того, что апоптоз гладкомышечных клеток играет роль ингибитора пролиферации [112, 113]. Этим можно объяснить повышенный фиброз и бедную клеточную архитектуру в более зрелых бляшках.
Следует подчеркнуть, что гладкомышечные клетки, мигрирующие в интиму, морфологически отличаются от нативных гладкомышечных клеток медии (рис. 9.24). Гладкомышечные клетки в атероматозной бляшке гистологически менее зрелые, но с высокой секреторной активностью. Эти клетки продуцируют соединительную ткань и клеточные компоненты, из которых образуется окружающий экстрацеллюлярный матрикс (ECM). Матрикс
|
PDGF |
IFN-γ |
|
Ag |
bFGF |
TGF-β |
|
|
IL-1 |
TNF-α |
|
|
TGF-β |
IL-1 |
|
|
PGI2 |
Т-лимфоцит |
|
|
IGF-1 |
|
bFGF |
|
NO |
|
|
|
|
IGF-I |
|
Клетка эндотелия ox LDL |
|
||
|
IL-1 |
||
|
|
|
M-CSF |
|
|
|
TGF-β |
|
|
Ag |
TNF-α |
|
|
|
PGE |
|
|
|
HB-EGF |
Плазма |
|
|
MCP-1 |
|
|
GM-CSF |
|
Ангиотензин |
|
|
|
|
|
Коллаген |
|
ЛНП |
|
Ag |
|
|
|
Эластичные волокна |
|
|
|
|
|
|
|
|
Протеогликан |
|
|
|
PDGF-AA |
|
Гладкомышечная клетка |
|
|
|
PDGF |
PDGF |
Ag |
|
EGF/TGF-α |
bFGF |
|
|
TGF-β |
HB-EGF |
|
|
TXA2 |
TGF-α |
|
Тромбоцит |
IGF-1 |
TGF-β |
|
TNF-α |
|
||
|
|
|
|
|
|
IL-1 |
Макрофаг |
|
|
PGE |
|
|
|
ox LDL |
|
Рис. 9.24. Два различных фенотипических состояния гладкомышечных клеток при атеросклерозе.
Исследованиями показано, что под воздействием циклической деформации гладкомышечные клетки увеличивают продукцию коллагена. Это накопление соединительной ткани повышает зрелость бляшки [114]. Кроме того, в процессе неоваскуляризации в области бляшки возникают микротромбы, которые служат очагами агрегации тромбоцитов и, следовательно, усиливают атеросклеротические изменения через PDGF-фактор. Как и на каждом предыдущем этапе образования бляшки, существуют регуляторные процессы, контролирующие образование ECM. Это достигается с помощью матриксных металлопротеиназ (MMPs), которые ферментативным путем расщепляют различные макромолекулы ЕСМ [115].
Период стабильной бляшки
При дальнейшем развитии бляшки может произойти ее разрыв, что вызывает острые симптомы. Неустойчивые бляшки более подвержены разрыву и последующему местному образованию тромбов. Богатые липидами бляшки более склонны к разрыву, чем бляшки, богатые коллагеном. Исследованиями выявлено, что повышенное содержание липидов, значительная воспалительная активность и сниженное заживление, связанное с деятельностью гладкомышечных клеток, являются теми факторами, которые определяют неустойчивость бляшек (табл. 9.4). Центральное ядро атеросклеротической бляшки лишено соединительной ткани и состоит главным образом из липидов и апоптических ядерных фрагментов [116–120]. Кроме того, склонные к разрыву бляшки содержат сложные эфиры холестерола в более высоких концентрациях, чем свободный холестерол [121, 122]. Участки бляшки, где фиброзная покрышка наиболее тонкая и наиболее сильно инфильтрирована пенистыми клетками, являются самыми слабыми к физическим воздействиям, которые приводят к их разрыву. Если размер ядра слишком увеличивается, это также повышает склонность к разрыву [123]. В новых исследованиях было показано, что пенистые клетки в высоких концентрациях обнаруживаются в местах разрыва фиброзной капсулы [124]. Иммунно-активные макрофаги способны к секреции MMPs, цистеиновых и серинных протеиназ, которые могут вызывать деградацию ЕСМ и фиброзной покрышки, что ведет к дальнейшей дестабилизации бляшки. Коллаген, секретируемый гладкомышечными клетками, служит для поддержания стабильности бляшки. Было обнаружено, что в области разрыва бляшки коллаген и неизмененные гладкомышеч-
Таблица 9.4. Факторы, влияющие на стабильность бляшки
Внутренние
1.Содержание липидов в атероматозном ядре
2.Циркулярное напряжение растяжения
3.Толщина фиброзной покрышки бляшки
4.Напряжение сжатия
Внешние
1.Воспаление покрышки бляшки
2.Напряжение изгиба
3.«Усталость» бляшки
4.Гемодинамические силовые воздействия