

Алемасова - Лекции по аналитической химии
.pdfХроматография – гибридный аналитический метод, в котором сочетаются метод разделения и метод определения.
B отличие от других методов, основанных на распределении компонентов между фазами, хроматография – это динамический метод, обеспечивающий многократность актов сорбции – десорбции разделяемых компонентов, так как разделение происходит в потоке подвижной фазы. Этим обусловлена большая эффективность хроматографического метода по сравнению с методами сорбции и экстракции.
Предложил хроматографию русский ботаник M.C. Цвет. Ha колонке, заполненной CaCO3, он разделял пигменты растений. Подвижной фазой служил диэтиловый эфир. Именно этим методом он обнаружил, что в экстракте зеленых листьев содержится 2 хлорофилла, 4 ксантофила и каротин. В табл. 4.1 представлена классификация хроматографических методов.
Таблица 4.1. Классификация видов хроматографии
Вид |
Подвижная |
Неподвижная |
Форма |
Механизм |
|
фаза |
фаза |
размеще- |
распределе- |
|
|
|
ния непод- |
ния |
|
|
|
вижной фа- |
|
|
|
|
зы |
|
ГАЗОВАЯ |
|
|
|
|
Газо-адсорб- |
газ |
твердая |
колонка |
адсорбци- |
ционная |
|
|
|
онный |
Газо- |
газ |
жидкость |
колонка |
распреде- |
жидкостная |
|
|
|
лительный |
ЖИДКОСТ- |
|
|
|
|
НАЯ |
|
|
|
|
Твердо- |
жидкость |
твердая |
колонка |
адсорбци- |
жидкостная |
|
|
|
онный |
|
|
|
|
|
Жидкость- |
жидкость |
жидкость |
колонка |
распреде- |
жидкостная |
|
|
|
лительный |
Ионообмен- |
жидкость |
твердая |
колонка |
ионный |
ная |
|
|
|
обмен |
ТОНКО- |
жидкость |
твердая |
тонкий |
адсорбци- |
СЛОЙНАЯ |
|
|
слой |
онный |
|
жидкость |
жидкость |
тонкий |
распреде- |
|
|
|
слой |
лительный |
БУМАЖНАЯ |
жидкость |
жидкость |
полоска |
распреде- |
|
|
|
бумаги |
лительный |
|
|
|
|
|
41
СИТОВАЯ |
жидкость |
жидкость |
колонка |
по |
разме- |
(ГЕЛЬ- |
|
|
|
рам |
моле- |
ПРОНИКА- |
|
|
|
кул |
|
ЮЩАЯ) |
|
|
|
|
|
|
|
|
|
|
|
Из данных табл. 4.1 видно, что классификация хроматографических методов проходит
–по агрегатному состоянию фаз;
–по механизму распределения;
–по технике выполнения.
B соответствием с режимом ввода пробы в хроматографическую систему, различают:
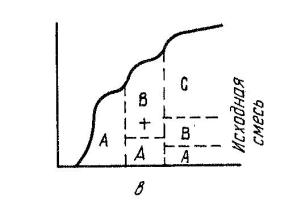
І. Фронтальная хроматография – если растворенную смесь непрерывно вводят в хроматографическую колонку, то в чистом виде можно выделить только одно, наиболее слабо сорбирующее вещество (рис. 4.1). Все остальные выйдут из колонки в виде смеси. Из колонки сначала будет вытекать чистый растворитель, затем, когда сорбент насытится менее сорбируемым веществом А, оно появится в элюате. Когда сорбент насытится вторым, менее сорбируемым веществом В, элюат будет содержать оба эти вещества и т.д. Когда же сорбент будет полностью насыщен всеми компонентами смеси, состав элюата совпадет с составом раствора, вводимого в колонку.
Сигнал детектора
время
Рис. 4.1. Фронтальная хроматограмма (сорбируемость веществ увеличивается в ряду A < B < C)
2. Элюентная – пробу вводят в поток подвижной фазы (элюента). Элюент (раствор или растворитель) обладает меньшей сорбируемостью, чем любое из разделяемых веществ. Затем в колонку вводят разделяемые вещества, растворенные в элюенте, и продолжают непрерывно пропускать элюент. При этом разделяемые вещества перемещаются вдоль колонки с разными скоростями в соответствии с их сорбируемостью. На выходе из колонки сначала появляется наименее сорбируемый компонент, затем следующий компонент и т.д. В этом случае хроматограмма представляет собой несколько пиков, имеющих форму гауссовой кривой (рис. 4.2).
42

время
Рис. 4.2. Элюентная хроматограмма (сорбируемость веществ увеличивается в ряду A < B < C)
3. Вытеснителъная - после введения пробы и предварительного разделения слабоактивным элюентом состав элюента меняется таким образом, что он взаимодействует с неподвижной фазой сильнее каждого из компонентов анализируемой смеси. Т.е. сначала в колонку вводят небольшое количество раствора разделяемых веществ. Затем через колонку непрерывно пропускают раствор вещества (вытеснителя), обладающего большей сорбируемостью, чем любое из разделяемых веществ. По мере продвижения по колонке элюент вытесняет вещество С, которое в свою очередь вытесняет вещество В, и т.д. В результате анализируемая смесь перемещается впереди фронта вытеснителя и скорость движения веществ равна скорости движения вытеснителя. Разделяемые вещества и на колонке, и в элюате располагаются последовательно друг за другом. Каждый из компонентов выделяется в чистом виде, но не количественно, так как зоны компонентов не разделены промежутками чистого сорбента (рис. 4.3).
Рис. 4.3. Вытеснительная хроматограмма (сорбируемость веществ увеличивается в ряду A < B < C)
Наиболее распространен элюентный режим, который позволяет получить в чистом виде все компоненты пробы.
43
В любой хроматографической системе происходит обратимый переход молекул вещества А из подвижной фазы (ПФ) в неподвижную фазу
(НФ)
Апф Ù Анф
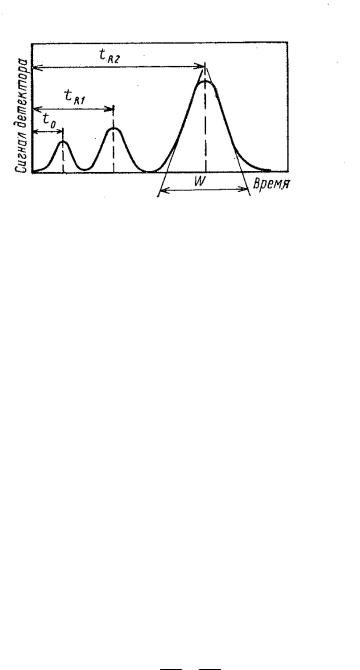
Хроматографический процесс характеризуется зависимостью концентрации вещества в элюенте от времени хроматографирования (хроматограмма), которая характеризуется следующими параметрами (рис. 4.4):
Рис. 4.4. Параметры хроматограммы: tR – время удерживания; W – ширина пика
Распределение веществ между двумя фазами характеризуется константой равновесия:
|
[Aнф |
] |
|
mнф |
Vпф |
Vпф |
|
|||||
K = |
|
|
= |
|
|
|
|
|
= k′ |
|
|
, |
[A |
] |
m |
пф |
V |
нф |
V |
нф |
|||||
|
пф |
|
|
|
|
|
|
|
|
|||
где k´—коэффициент емкости; Vпф и Vнф - объем подвижной и неподвижной фаз; mпф и mнф – количество вещества в подвижной и неподвижной фазах
Время выхода (удерживания) соответствующей зоны из колонки tR связано с коэффициентом емкости :
k |
′ |
= |
tR −t0 |
, |
|
t0 |
где tR – время удерживания компонента А; t0 – время выхода вещества, не взаимодействующего с неподвижной фазой (мертвое время или время прохождения через колонку несорбируемого компонента).
Различие во временах удерживания компонентов характеризует селективность системы, выражаемую величиной α:
α= k'2 = K2 k'1 K1
Таким образом, α равно отношению коэффициентов равновесного распределения К или коэффициентов емкости k′ 2-х компонентов и яв-
ляется мерой термодинамического различия в их распределении:
∆G0 = – RT·ln α,
44

где ∆G0 – различие в свободной энергии сорбции 2-х компонентов.
B процессе движения по колонке зона вещества вследствие диффузии размывается, что сказывается на ширине пиков. Ширина пиков определяется эффективностью хроматографической системы: на представленной ниже хроматограмме в первом случае разделение двух веществ довольно селективно, но неэффективно; во втором случае – высокая эффективность и высокая селективность разделения.
I II
В качестве меры размывания хроматографической полосы используют параметр H, имеющий размерность длины и называемый “высота, экви-
валентная теоретической тарелке” (ВЭТТ):
H = L / 16·(W/tR)2,
где W – ширина пика; L - длина колонки.
W
Чем меньше H, тем уже пик, тем эффективнее система и большее количество компонентов можно разделить на колонке.
Теория теоретических тарелок
Хроматографическую колонку можно представить, как ряд дискретных, несоприкасающихся узких слоев или зон – “тарелок”. На каждой тарелке устанавливается равновесное распределение вещества. Движение подвижной фазы приводит к переносу части вещества с одной тарелки на другую и далее.
Время удерживания tR пропорционально числу теоретических тарелок:
tR = b·N,
где b – коэффициент пропорциональности; N – число теоретических тарелок.Для расчета N можно использовать параметр tR:
N=16 tR 2
W
Зная число теоретических тарелок и длину колонки, можно рассчитать
H:
Н = NL
45
Диффузная (кинетическая) теория хроматографии
По этой теории принимается во внимание, что в динамических условиях на размывание хроматографической зоны влияет диффузия молекул сорбата в подвижной фазе, в порах сорбента и другие сложные процессы массообмена. Связь ВЭТТ (H) с линейной скоростью (U) потока подвижной фазы описывается уравнением:
Н= A + UB +C U
Вэтом уравнении коэффициентом А выражают вихревую диффузию;
В– коэффициент диффузии, связанный с движением подвижной фазы вдоль колонки; коэффициент С выражает сопротивление массопередачи в двух фазах, U – скорость потока подвижной фазы.
При больших скоростях потока подвижной фазы в жидкостной адсорбционной хроматографии (“высокоскоростная хроматография”) величина Н зависит в основном от слагаемых А и С·U в представленном выше уравнении, поэтому увеличение скорости U приводит к непрерывному увеличению Н, т.е. к уменьшению эффективности работы хроматографической колонки.
Для проведения хроматографических измерений используют хроматограф. Основными узлами хроматографов являются дозатор (система ввода пробы), хроматографическая колонка и детектор. Кроме того, в хроматографе имеются устройства для подачи газа-носителя или растворителя, для преобразования импульса детектора в соответствующий сигнал и некоторые другие.
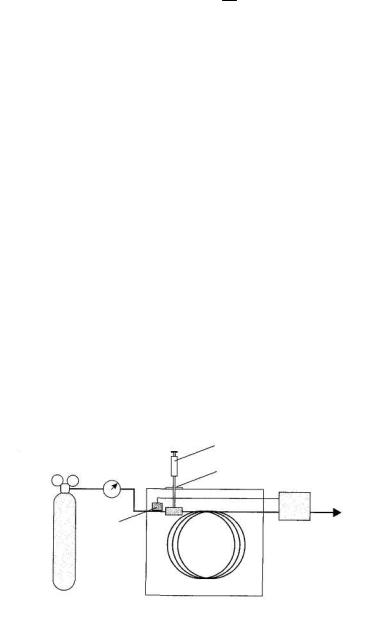
Устройство газового хроматографа может быть представлено следующим образом (рис. 4.5):
1
2
6
3
4 |
5 |
Рис. 4.5. Блок-схема газового хроматографа:
1 – газовый шприц для ввода образца; 2 – мембрана; 3 – обходной клапан; 4 – баллон с газом-носителем; 5 – колонка; 6 – детектор Газообразные и жидкие пробы обычно вводят с помощью специаль-
ных шприцев, прокалывая в месте ввода пробы каучуковую мембрану. В хроматографической колонке происходит разделение компонентов. При-
46
меняют прямые, спиральные и другие колонки длиной от 1–2 м и менее до нескольких десятков метров. Колонки бывают металлические, стеклянные, пластиковые. Колонок и детекторов может быть несколько. В газовой хроматографии в качестве адсорбентов используют оксид алюминия, силикагели, активированные угли, пористые полимеры на основе стирола, дивинилбензола и т.д. и синтетические цеолиты. Обязательно компьютер и банк хроматографических данных. Сейчас используют капиллярные колонки из плавленного кварца. Большое влияние на сорбируемость вещества оказывает температура, поэтому хроматографические колонки, как правило, термостатируются.
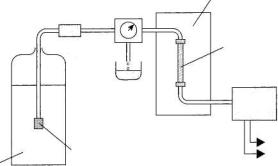
Приборы для жидкостной хроматографии имеют те же принципиальные узлы. На рис. 4.6 представлена схема хроматографа для высокоэффективной жидкостной хроматографии (ВЭЖХ).
4
3
5
6
2
1
Рис. 4.6. Жидкостный хроматограф для ВЭЖХ:
1 – подвижная фаза; 2 – фильтр; 3 – насос; 4 – термостат; 5 – колонка; 6 – детектор
Насос по определенной программе подает элюент в хроматографическую колонку. Для обеспечения высокой скорости анализа насосы создают давление до 40 МПа. Проба через специальное устройство (инжектор) вводится непосредственно в поток элюента. После прохождения через хроматографическую колонку вещество детектируется проточным детектором.
Хроматограмма, представленная на рис. 4.4, является носителем качественной и количественной информации. Так, время удерживания tR – качественный параметр. Совпадение величин времени удерживания неизвестного и стандартного соединений свидетельствует о том, что эти соединения могут быть идентичными. Площадь под кривой и нулевой линией детектора (площадь хроматографического пика) S или высота хроматографического пика h являются количественными параметрами.
Cпособы количественного анализа
1. Метод нормировки
Для его использования необходимо, чтобы на хроматограмме были зарегистрированы все компоненты, входящие в состав анализируемой смеси. Доля площади пика соответствует содержанию компонента в массовых
47
процентах. При анализе смеси 3х компонентов содержание компонента, например, соответствующего пику х на хроматограмме, можно рассчитать по формуле
x,% = |
Sx |
100% |
Sx + S y + Sz |
Эту формулу используют только в том случае, если детектор одинаково чувствителен к каждому из разделяемых компонентов смеси. Если же чувствительность детектора различна по отношению к каждому из компонентов пробы, то используют поправочные коэффициенты fx, fy, fz, учитывающие чувствительность детектора к данному компоненту. Формула для расчета в этом случае записывается так:
x,% = |
S |
f |
100 |
x |
x |
||
∑S |
n fn |
Коэффициент fx находят при анализе стандартных веществ. Для этого готовят смесь точно определенных количеств (по массе и объему) исследуемых компонентов. Модельную смесь хроматографируют, находят пло-
щади пиков и рассчитывают поправочные коэффициенты fi = mi
Si
2. Метод внешнего стандарта
Метод используют при определении отдельных веществ или анализе простых смесей, а также микропримесей. Готовят два стандартных раствора определяемых компонентов, одинаковые их количества вводят в хроматограф и определяют площади пиков (или высоту) каждого компонента S1, Sx и S2. Далее по двум ограничивающим точкам или по большему числу точек строят зависимости площади пика от содержания определяемого компонента в стандартных растворах (градуировочный график).
3. Метод внутреннего стандарта
Его применяют при отсутствии на хроматограмме пиков некоторых компонентов анализируемой смеси. B анализируемую смесь вводят некоторое количество стандартного вещества. Это вещество должно быть химически инертным, отсутствовать в анализируемой пробе и полностью отделяться от других компонентов смеси. Время его удерживания должно быть близким к tR определяемых компонентов. Его концентрация должна быть близка к концентрациям определяемых компонентов.
Sx |
Sy |
|
|
Sz |
|
|
|
|
|
|
Sвн.ст. |
48

Определяют площади пиков и для каждого компонента рассчитывают поправочный коэффициент по формуле
k = Sвн.ст. Сх
Sx Cвн.ст.
Зная поправочные коэффициенты, содержание компонента рассчитывают по формуле
mвн.ст. |
|
|
|
|
|
|
|
|
Sx |
|
, |
||||
х,% = k |
m |
|
|
|
|
100 |
|
|
|||||||
|
пробы |
|
Sвн.ст. |
|
|
||
где mвн.ст. и mпробы – масса соответственно внутреннего стандарта и пробы, г.
4.2. Жидкостная хроматография
Подвижная фаза – жидкость. B методе жидкостной хроматографии разделение чаще всего происходит при комнатной температуре. B отличие от газа, который выполняет только транспортную функцию и не сорбируется подвижной фазой, жидкая подвижная фаза - активный элюент, молекулы которого могут сорбироваться на поверхности. При прохождении через колонку находящиеся в элюенте молекулы интересующего нас компонента должны вытеснить молекулы элюента с поверхностного слоя сорбента.
Селективность в жидкостной хроматографии, в отличии от газовой, определяется не одним, а двумя факторами – природой подвижной (элюент) и неподвижной фаз.
B стеклянную колонку длиной 1-2 м, заполненную сорбентом, вводят анализируемую пробу и пропускают элюент. Размер зерен сорбента 5-30 мкм. Для таких сорбентов необходимо принудительное нагнетание элюента через колонку. Этот метод называется высокоэффективная жидкостная хроматография (ВЭЖХ). Метод может использоваться для разделения молекул (твepдo-жидкocтная и жидкость-жидкостная хроматография), для разделения и определения ионов (ионообменная, ионная, ион-парная), для разделения макромолекул (эксклюзионная).
4.2.1. Адсорбционная хроматография
Неподвижная фаза должна удерживать разделяемые вещества. Подвижная фаза, т.е. растворитель, должна обеспечивать различную емкость колонки и эффективное разделение за приемлемое время.
Неподвижные фазы – это адсорбенты различных типов (полярные, неполярные, пористые носители). Это тонкодисперсные пористые материалы.
Полярные адсорбенты (SiO2, A12O3, оксиды металлов, флоросил). Все эти адсорбенты имеют на поверхности слабокислотные OH - группы,
49
способные удерживать вещества с основными свойствами. Эти адсорбенты применяют для разделения неполярных соединений и соединений со средней полярностью. Недостаток полярных адсорбентов - высокая чувствительность к содержанию H2О в растворах, например, силоксановые группы −Si – O – Si – на поверхности SiO2 в присутствии воды переходят в силанольные ≡ Si – OH.
Неполярные адсорбенты - графитовая сажа, кизельгур, диатомит. Используют также сорбенты с привитыми неполярными фазами, например, силикагель с алкилсилильными группами от С2 до С22.
Кроме того, используют поверхностно-пористые носители – стеклянные шарики, покрытые тонким пористым слоем активного полярного или неполярного сорбента.
Подвижные фазы:
Подвижная фаза должна растворять анализируемую пробу, обладать малой вязкостью, должны быть инертной по отношению к материалам всех частей хроматографа.
Для SiO2 растворители – CC14, С6Н6, CHC13, диоксан, ацетонитрил. Применяют пропанол, изопропанол, этанол. Часто применяют не индивидуальный растворитель, а их смесь. Иногда применяют метод ступенчатого или градиентного элюирования, т.к. одна подвижная фаза в качестве элюента может не разделить все компоненты пробы за приемлемое время.
Механизм удерживания чаще всего смешанный, т.е. удерживание происходит по адсорбционному, распределительному, эксклюзионному механизмам.
4.2.2. Распределительная хроматография
Метод распределительной или жидкость – жидкостной хроматографии основан на распределении вещества между двумя несмешивающимися жидкостями. Жидкая неподвижная фаза наносится на пористый инертный сорбент, как в газожидкостной хроматографии, и им заполняют разделительную колонку. При пропускании жидкой подвижной фазы через колонку смесь разделяется на компоненты за счет их различной растворимости в жидкой неподвижной фазе и в основном по тем же механизмам, что и в газожидкостной хроматографии.
Обычно полярный растворитель (H2О, спирт) фиксирован на твердом носителе – силикагеле SiO2, целлюлозе, диатомите, оксиде алюминия A12O3. Подвижной фазой в этом случае служат неполярные растворители - изооктан, С6Н6. Если на носителе зафиксирован неполярный растворитель, то в качестве подвижной фазы используют полярные растворители (воду, спирт, буферные растворы, сильные кислоты). Haнeceнные жидкие фазы имеют большой недостаток. Они быстро смываются подвижной жидкой
50