

3 курс / Патологическая физиология / Сахарный диабет
.pdfКафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
ГЛАВА 12. ПАТОФИЗИОЛОГИЯ УГЛЕВОДНОГО ОБМЕНА
Уровень глюкозы в крови является одним из наиболее важных гомеостатических параметров в организме.
Он регулируется различными органами-системами (например, ЦНС, печенью, кишечником, почками и т.д.) бла-
годаря их взаимосогласованной деятельности и, прежде всего, эндокринной системе. Биологической целью под-
держания этого показателя является обеспечение мозга практичски единственным субстратом энергии. Прием пищи приводит к высвобождению многих гормонов, которые регулируют гомеостаз глюкозы и чувство на-
сыщения. Гормоны, регулирующие обмен глюкозы, место их синтеза и их эффекты, представлены в таблице 12.1.
Таблица 12.1.
Гормон |
Место выработки |
Действие |
|
|
|
глюкагон |
α-клетки островков Лангерганса |
Стимулирует глюконеогенез и гликогенолиз. |
|
|
|
глюкокортикоиды |
кора надпочечников |
Активируют глюконеогенез и угнетают активность |
|
|
гексокиназы. |
|
|
|
катехоламины |
мозговое вещество надпочечников |
Активируют гликогенолиз. |
|
|
|
тиреоидные гормоны |
щитовидная железа |
Активируют гликогенолиз, глюконеогенез, всасывание |
|
|
глюкозы в кишечнике, угнетают гликогенез |
|
|
|
СТГ |
аденогипофиз |
Активирует гликогенолиз, инсулиназу в печени, |
|
|
угнетает утилизацию глюкозы тканями. |
|
|
|
инсулин |
β-клетки островков Лангерганса |
Снижает количество глюкозы в крови. |
|
|
|
инкретины |
тонкий кишечник |
Стимулируют высвобождение инсулина. |
|
|
|
Одной из важнейших групп гормонов являются инкретины, которые ответственны за стимуляцию высво-
бождения инсулина панкреатическими β-клетками после приема пищи. Двум из которых придается особое зна-
чение: глюкозозависимому инсулинотропному полипептиду (GIP: glucose-dependent insulinotropic polypeptide) и
глюкагоноподобному пептиду-1 (GLP-1: glucagon-like peptide-1). После приема пищи в крови увеличивается содержание GIP и GLP-1, это явление называется «инкретиновым эффектом». Помимо воздействия на высвобож-
дение инсулина, под действием инкретинов снижается секреция глюкагона, замедляется эвакуация содержимого желудка, что стимулирует формирование чувства насыщения. Циркулирующие инкретины в дальнейшем инак-
тивируются ферментами семейства дипептидилпептидаз (DPP), важное значение придается ферменту DPP-4.
Содержание глюкозы в крови колеблется в пределах 3,58-6,05 ммоль/л. Состояния, характеризующиеся понижением уровня глюкозы в крови (ниже 3,58 ммоль/л), называются гипогликемическими.
По происхождению различают наследственные или первичные и вторичные или симптоматические гипогликемии. Перивчные гипогликемии могут быть следствием нарушения разрушения гликогена (например,
при гликогенозах), или его депонирования (например, при дефиците или блокаде гликогенсинтетазы). По своему биологическому значению различают физиологическую и патологическую вторичные гипогликемии. Развитие физиологической гипогликемии возможно в следующих случаях: тяжелая и продолжительная физическая или психическая нагрузка, в период лактации у женщин, сразу после алиментраной гипергликемии и т.д.
Патологическая гипогликемия может быть обусловлена гиперинсулинизмом, например, при инсуломе.
Такая гипогликемия чаще развивается у больных сахарным диабетом из-за передозировки инсулина. Существуют также гипогликемии без гиперинсулинизма, например, подобное состояние наблюдается при надпочечниковой недостаточности (дефицит глюкокортикоидов), при различных поражениях канальцевого аппарата почек.
1
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
Основой развития почечной гипогликемии является снижение почечного порога для глюкозы, в результате развивается глюкозурия, которая приводит к гипогликемии.
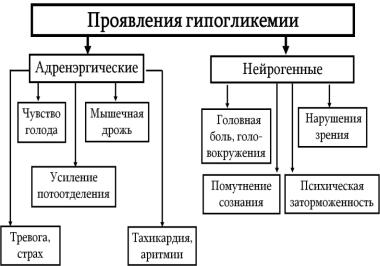
Проявления гипогликемии представлены на рисунке 12.1.
Состояния, характеризующиеся повышением уровня глюкозы в крови (выше 6,05 ммоль/л)
называются гипергликемическими. Следует отметить, что гипергликемические состояния встречаются чаще, чем гипогликемические.
По биологическому значению различают фи-
зиологические и патологические гипергликемии.
Физиологическая гипергликемия является кратко-
временным и самостоятельно обратимым состоя-
нием. Физиологическая гипергликемия является кратковременным и самостоятельно обратимым состоянием. Развитие алиментарной гипергликемии
связано с приемом пищи богатой углеводами.
Нейрогенная же гипергликемия развивается при психо-эмоциональном стрессе и обусловлена метаболическим воздействием стресс-гормонов.
Патологические гипергликемии могут развиваться также в голодном состоянии. В основе развития
нейроэндокринной гипергликемии лежит нарушение оптимального взаимоотношения в крови гормонов с гипо-
и гипергликемическими воздействиями, например, при феохромоцитоме, опухолях коры надпочечников, и т.д.
Развитие судорожной гипергликемии обусловлено стимуляцией гликогенолиза во время судорог. Образуется лактат, который превращается в глюкозу в печени. Гипергликемии, разивающиеся вследствие употребления наркотиков, например кокаина, связаны с активацией СНС.
12.1.САХАРНЫЙ ДИАБЕТ
Кгипергликемическим состояниям относится сахарный диабет (СД) (diabetes mellitus: от греч. diabetes -
перехожу, пересекаю и лат. mellitus - мед). Термин «Диабет» был введен в литературу примерно в 20г. н.э.
Арсением Каппадокийским, а «Сахарный диабет» - Томасом Уилисом (1679).
Еще в Xв. Авиценна описал ряд признаков СД: постоянное чувство жажды, полиурия, повышение аппетита,
быстрая утомляемость, наличие в моче сладкого осадка («меда»).
В 1889г. Меринг и Минковский выяснили, что после удаления у собаки поджелудочной железы у нее развиваются нарушения углеводного обмена: гипергликемия, глюкозурия и другие явления, которые напоми-
нают СД у людей. При трансплантации части удаленной железы нарушения обмена постепенно исчезают.
В 1894г. английский ученый Эдвард Шафер выдвинул предположение, что гормон, вырабатываемый островками Лангерганса регулирует обмен глюкозы в организме. Он назвал этот гормон инсулином (insula
означает островок).
В 1900-1901гг. Соболев провел блестящий эксперимент, перевязав проток поджелудочной железы, показав,
что несмотря на то, что ацинарная ткань подвергается атрофии, диабет у животного не развивается (островки Лангерганса сохранены). Таким образом, Соболев пришел к выводу, что морфологическим субстратом эндокринной деятельности поджелудочной железы являются островки Лангерганса.
2

Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
В 1922г. научной группе канадского хирурга Фредерика Ф. Бантинга впервые удалось выделить инсулин.
Бантинг пытался выделить инсулин из поджелудочной железы. Многие научные исследования оказались безуспешными, поскольку протеолитические ферменты железы, в частности трипсин, разрушали гормоны поджелудочной железы. Вместе со своим молодым помощником, Чарльзом Бестом, он перевязал панкреатические протоки собак, таким образом подвергая дегенерации ацинусы поджелудочной железы.
Фактически для выделения инсулина он моделировал панкреатит. В этом случае жизнеспособными оставались только клетки инсулинового аппарата, и инсулин, выделенный из экстракта железы, не разрушался.
Руководитель лаборатории, физиолог Джон МакЛеод, создавший необходимые условия для экспериментов,
позже присоединил к группе ислледователей биохимика Джеймса Колипа. Последний очищал экстракт инсулина, до той степени, чтобы его можно было вводить детям с тип I диабетом, избегая осложнений. В 1923
году Бантинг и МакЛеод были удостоены Нобелевской премии за выделение инсулина. Бантинг отдал половину своей премии своему младшему другу Бесту, а МакЛеод - Колипу.
В1926-1927 гг. Абель и Скотт получили кристаллический инсулин. В конце 1963 г. Кацояннис (США), Зан
иМейенхофер (Германия) синтезировали инсулин.
Сахарный диабет - группа гетерогенных заболеваний, характеризующихся гипергликемией, в основе развития которых лежит абсолютная и/или относительная недостаточность инсулина.
Сахарный диабет является серьезной медицинской и социальной проблемой, о чем свидетельствуют приведенные данные.
СД занимает первое место среди эндокринопатий.
СД является третьей или четвертой причиной смерти человека.
1-2% населения планеты страдают СД, и по данным ВОЗ наблюдается тенденция увеличения заболеваемости диабетом.
Каждые 10-15 лет число больных СД удваивается.
Расходы на лечение больных СД в развитых странах составляют 10-15% от всех расходов здравоохранения.
10% людей в возрасте 70 лет страдают тип II СД.
По данным ВОЗ у лиц с избыточным весом тела заболеваемость СД увеличивается в 5-10 раз.
На периферии островков Лангерганса поджелу-
дочной железы расположены -клетки, продуциру-
ющие глюкагон, а в центральной части β-клетки,
продуцирующие инсулин (рис. 12.2). В островках расположены также δ-клетки, составляющие до 5%, и
которые продуцируют соматостатин.
Молекула инсулина состоит из двух полипептидных цепей: A (21 аминокислот) и B (30
аминокислот), которые соединены дисульфидными мостиками. Молекула инсулина склонна к обратимой агрегации.
Рис. 12.2. Клеточный состав островков Лангерганса. |
|
В |
-клетках |
осуществляется |
биосинтез |
||
|
|
|
|
|
|||
|
инсулина |
в |
следующей |
последовательности: |
|||
рибосомы синтезируют препроинсулин (состоящий из 110 аминокислот), который под действием специфической пептидазы превращается в проинсулин, состоящий из 86 аминокислот. Последний накапливается в β-гранулах.
Далее, под действием специфической пептидазы от него отделяется С-пептид и образуется инсулин, который
3
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
легко полимеризуется: два атома цинка соединяют 6 молекул инсулина, образуя гексамер. Последний в виде кристаллов содержится в секреторных гранулах островков поджелудочной железы.
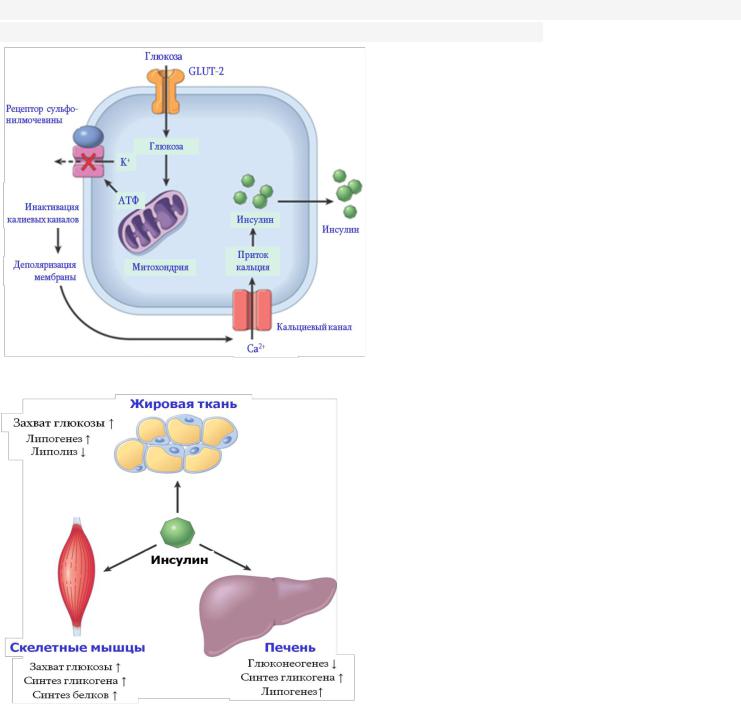
Известно, что глюкоза является основным стиму-
лятором секреции инсулина β-клетками. Проникая в β-
Проникая в β-
клетки посредством инсулинезависимого транспортера
GLUT-2, глюкоза способствует усилению синтеза АТФ,
которая связывается с калиевыми канальцами, инакти-
вируя последние (рис. 12.3). В результате уменьшается выход калия из клеток, вследствие чего мембрана де-
поляризуется, что приводит к открыванию потенциал-
зависимых Ca-каналов. Повышенное поступление
|
|
|
кальция в клетку стимулирует выделение инсулина. |
|
|
|
|
|
Высвобождение инсулина также стимулируют n. |
|
|
|
vagus, аминокислоты, жирные кислоты и др. Его |
|
|
|
|
высвобождение подавляется гипогликемией, адренали- |
|
|
|
|
ном, соматомедином (путем угнетения высвобождения |
|
|
Рис.12.3. Регуляция высвобождения инсулина. |
СТГ). |
||
|
|
|
|
Инсулин расщепляется в тканях, в особенности в |
|
|
|
печени, под действием протеолитического фермента - |
|
|
|
|
инсулиназы. Биологическое значение последнего |
|
|
|
|
заключается в предотвращении чрезмерного накопления |
|
|
|
|
инсулина в организме и, тем самым, развитие |
|
|
|
|
гипогликемии. |
|
|
|
|
|
|
|
|
|
|
Чтобы понять влияние инсулина на основные ми- |
|
|
|
|
|
|
|
|
шени, необходимо помнить, что это важный анаболичес- |
|
|
|
|
|
|
|
|
|
кий гормон, который стимулирует синтез жиров из жир- |
|
|
|
|
|
|
|
|
|
ных кислот, синтез гликогена из глюкозы, синтез белков |
|
|
|
|
|
|
|
|
|
из аминокислот (рис. 12.4). С другой стороны, инсулин |
|
|
|
|
|
|
|
|
|
делает все возможное, чтобы снизить уровень глюкозы в |
|
|
|
|
|
|
|
|
|
крови: усиливает экспрессию переносчиков глюкозы |
|
|
|
|
|
|
|
|
|
GLUT4 (усиливая захват глюкозы) в мышцах и жировой |
|
|
|
|
|
|
|
|
|
ткани, стимулируя синтез гликогена или жиров, |
|
Рис. 12.4. Мишени воздействия инсулина. |
|
|||
облегчает утилизацию глюкозы в организме. Помимо |
||||
|
|
|
|
|
|
|
|
упомянутых эффектов, инсулин в печени блокирует |
|
|
|
|
|
|
|
гликонеогенез и гликогенолиз, подавляет кетогенез. |
|
|
|
12.1.1. КЛАССИФИКАЦИЯ САХАРНОГО ДИАБЕТА
Международная федерация диабета в 2009г. приняла следующую классификацию:
1. тип I диабет:
а) аутоиммунный,
б) идиопатический, 2. тип II диабет,
4
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
3.сецифические формы диабета:
•генетические дефекты функционирования β-клеток,
•генетические дефекты действия инсулина (например, тип А инсулинорезистентность),
•поражения «экзокринной поджелудочной железы» (например, хронический панкреатит, травма),
•эндокринопатии (например, синдром Кушинга, акромегалия, феохромоцитома и т.д.),
•инфекции (например, цитомегаловирус, врожденная краснуха и т.д.),
•лекарства (например, тиазиды, глюкокортикоиды и т.д.),
•диабет -ассоциированные генетические синдромы (например, синдром Дауна и т.д.),
4.диабет беременных.
Общим признаком всех видов сахарного диабета является гипергликемия, однако ее механизмы развития различны. Современную классификацию СД можно назвать патогенетической, так как она наилучшим образом отражает механизмы, лежащие в основе развития различных видов диабета.
11.1.2. ЭТИОЛОГИЯ И ПАТОГЕНЕЗ ТИП I ДИАБЕТА
Основным патогенетическим звеном данного диабета является аутоиммунное воспаление, приводящее к деструкции β-клеток. В результате, из-за уменьшения общего количества β-клеток развивается абсолютная недостаточность инсулина. Такие больные полностью зависят от экзогенного введения инсулина, а при его
|
отсутствии развиваются метаболические осложнения: |
|||
|
кетоацидоз и кома. |
|
|
|
|
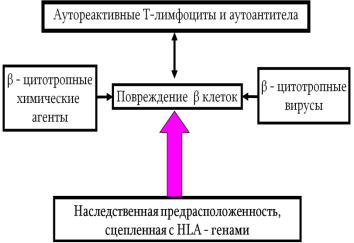
Развитие аутоиммунных реакций к β-клеткам обус- |
|||
|
ловливает наличие дефекта локуса HLA-D в 6-й |
|||
|
хромосоме. Эти реакции развиваются спонтанно или, что |
|||
|
более вероятно, у лиц с генетическими дефектами |
|||
|
запускаются под действием различных внешних |
|||
|
факторов (вирусов, химических веществ и т.д.) и |
|||
|
приводит к аутоиммунному воспалению островков |
|||
|
Лангерганса: инсулиту и повреждению β-клеток |
|||
|
компонентами иммунной системы (рис. 12.5). |
|||
|
Генетические |
нарушения экспрессируют |
избыточное |
|
Рис. 12.5. Этиология тип I сахарного диабета |
количество |
DR-антигенов. |
Приблизительно у 95% |
|
(по А.В. Атаман, с нашими дополнениями). |
пациентов с тип I диабетом выявляется DR3 и/или DR4. |
|||
|
У людей с |
упомянутыми |
антигенами |
вероятность |
развития СД в 3 раза выше, чем при отсутствии этих антигенов. Вероятность развития заболевания увеличивается в 8-10 раз при сочетании антигенов DR3+DR4. Болезнь ассоцируется также с антигенами B-сублокуса (B8, B15).
Следует отметить, что аутоиммунная деструкция имеет длительно скрытое течение, и лишь при гибели 80-
90% проявляется клинически. В пользу аутоиммунного механизма свидетельствуют следующие факты: у
примерно 90% пациентов с тип I диабетом обнаруживаются маркеры иммуной деструкции β-клеток: антитела против антигенов β-клеток (ICA), инсулина (IAA), декарбоксилазы глютаминовой кислоты (GAD),
тирозинфосфатаз (IA2 и IA2b). У 10% этих пациентов выявляются и другие аутоиммунные заболевания, такие как болезнь Аддисона, Грейвса, тиреоидит Хашимото, целиакия и другие. И, наконец, иммуносупрессивная терапия у экспериментальных животных и детей предотвращает развитие заболевания или облегчает ее течение.
5
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
Давно известно, что СД является семейным заболеванием. Однако конкретный механизм наследования дефектных генов, ответственных за развитие тип I СД, остается неизвестным. Конкордантность у монозиготных близнецов (т.е. проявление генетической специфики у обоих близнецов) составляет 50%. У детей, родители которых страдают тип I диабетом, заболеваемость составляет лишь 5-10%.
Как уже упоминалось, аутоиммунная реакция может быть спровоцирована внешними факторами и, прежде всего, вирусами. Чаще всего это возбудители эпидемического паротита, кори и болезни Коксаки. Причем вирус Коксаки имеет даже диабетогенный штамм. Однако инфицированность этим вирусом приближается к 50%, а
заболеваемость диабетом состовляет 4%. Вирусы могут индуцировать развитие диабета тремя механизмами. Пер-
вый неспецифический механизм заключается в повреждении вирусом клеток островков Лангерганса и индуци-
рование воспаления, что приводит к высвобождению β-клеточных антигенов и активации аутореактивных Т-кле-
ток. При втором механизме вирусы продуцируют белки, похожие на β-клеточные антигены (молекулярная ми-
микрия). При иммунной реакции на них повреждается и собственная ткань. Третий механизм, заключается в том,
что перенесенные ранее вирусные инфекции могут персистировать в некоторых тканях и спустя некоторое время вызвать реинфекцию с высвобождением вируса с антигенными детерминантами. Это вызывает иммунный ответ на инфицированную клетку. Этот механизм известен как «вирусное дежавю». Несмотря на все перечисленное,
еще не до конца ясно по какому из этих механизмов происходит повреждение β-клеток, а также неизвестна вирусная инфекция, которая является причиной вышеуказанного.
В ответ на высвобождение антигенов поврежденными клетками островков Лангерганса активируются аутореактивные Т-клетки, которые повреждают β-клетки. В процессе участвуют разные популяции Т-клеток, в
том числе клетки TH1, которые повреждают β-клетки, вырабатывая γ-интерферон и ФНО, а также цитотоксичес-
кие CD8+ Т-лимфоциты, которые непосредственно уничтожают β-клетки. Мишенями иммунных реакций являются инсулин, β-клеточный фермент декарбоксилаза глютаминовой кислоты (GAD) и 512 аутоантиген клеток островков Лангерганса (ICA 512).
У больных диабетом в островках поджелудочной железы нередко выявляются довольно интенсивные лимфоцитарные инфильтраты. В инфильтратах выявляются CD4+ и CD8+ Т-клетки. Такая картина наблюдается в экспериментальных моделях тип I диабета. Причем, при введении клеток CD4+ от больных животных к здоровым, у последних развивается диабет. Выяснено, что часть этих CD4+ T-клеток являются TH17, которые способствуют миграции лейкоцитов в островки, активацию моноцитов (макрофагов) и развитию местного
иммунного воспаления. Было также показано, что такое агрессивное отношение к собственным клеткам является результатом недостаточности специфичных Treg.
В патогенезе тип I диабета также участвуют антитела. Синтез антител в основном стимулирует-
ся под действием фоликулярных Т-хелперов (TFH).
Выработанный ими ИЛ-21 способствует синтезу
IgG с высокой афинностью к указанным антигенам.
Более того, их наличие является прогностическим маркером заболевания, однако неясно, являются ли антитела причиной или следствием повреждения.
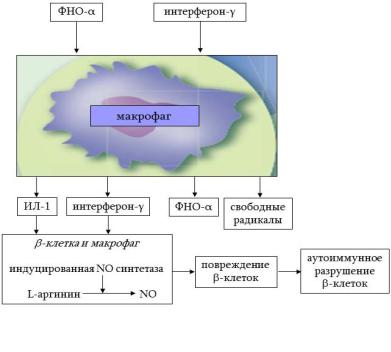
Имунные реакции, приводящие к деструкции
β-клеток, интерферон-γ и ФНО активируют макрофаги островкового аппарата. Они, в свою
6
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
очередь, высвобождают интерферон-γ, ФНО-α, ИЛ-1, свободные радикалы, которые усиливают процесс разруше-
ния β-клеток. Кроме того, ИЛ-1 и интерферон-γ стимулируют экспрессию индуцибельной (iNOS) NO-синтетазы в β-клетках и макрофагах (рис. 12.6). Под действием NO-синтетазы из L-аргинина образуется NO, который играет значительную роль в механизмах развития сахарного диабета, оставляя цитотоксический и цитостатический эффект. Важным патогенетическим механизмом тип I диабета является запуск β-клеточного апоптоза. Последний возникает как защитный механизм при внедрении тропных вирусов в клетки. Таким образом, β-клетки «извне» повреждаются вследствие разрушительного действия компонентов иммунной системы, а «изнутри» - в результате атаки NO.
Таким образом, учитывая вышеизложенное, можно определить, что тип I диабет является органоспецифическим аутоиммунным заболеванием.
Подводя итоги, патогенез сахарного тип I диабета можно представить как следующую последовательность событий: генетическая предрасположенность - провоцирующее влияние факторов внешней среды – инсулит -
аутоиммунная активация - иммунная атака на β-клетки - разрушение более 90% β-клеток - сахарный диабет.
Причины некоторых случаев тип I диабета остаются невыявленными и они называются идиопатическим диабетом. У этих пациентов наблюдается постоянный дефицит инсулина и предрасположенность к кетоацидозу.
У них отсутствуют маркеры иммуного разрушения и связь с генами HLA. Идиопатический диабет часто развивается у представителей африканского и азиатского происхождения, что, вероятно, связано с частым употреблением продуктов, богатых цианидами.
12.1.3. ЭТИОЛОГИЯ И ПАТОГЕНЕЗ ТИП II ДИАБЕТА
Это классический пример полиэтиологической болезни. Этиология и патогенез этой формы диабета менее изучены. Патогенетической основой тип II диабета является инуслинорезистентность и дисфункция β-клеток
(относительная недостаточность инсулина).
Наследственная предрасположенность играет более важную роль при тип II диабете, чем при тип I диабете.
Так, конкордантность у монозиготных близнецов выше 90%. Если один из родителей страдает этим типом диабета, то в 40% случаев дети наследуют это заболевание. А если болеют оба родителя, вероятность того, что дети заболеют, увеличится в два раза. Однако, в отличие от тип I диабета, болезнь не связана с генами HLA и
аутоиммунными механизмами. Проявлению наследственной предрасположенности к тип II СД способствует образ жизни приводящее к ожирению: переедание и низкая физическая активность. Типа II диабет является ключевым компонентом «метаболического синдрома».
Механизмы наследования тип II сахарного диабета и локализация генетических дефектов неизвестны. Как было уже отмечено, в основе развития тип II диабета лежит снижение чувствительности тканей к инсулину и способности отвечать на его воздействие - инсулинорезистентность (ИР).
В основе развития ИР могут лежать нарушения связывания инсулина с рецепторами (рецепторный механизм ИР), а также пострецепторной сигнализации. Вследствие нарушения последней в мембранах клеток-
мишеней угнетается экспрессия GLUT-4, нарушается захват глюкозы из крови, что приводит к развитию гипергликемии. Нарушаются также и другие эффекты инсулина: синтез гликогена, липогенез и т.д.
Для понимания рецепторных и пострецепторных механизмов следует отметить, что нормальная сигнализация инсулиновых рецепторов подразумевает тирозиновое фосфорилирование цитоплазматического сегмента рецептора (Tyr-P на рис. 12.7). Напротив, сериновое фосфорилирование (Ser-P) блокирует активность рецептора. Следовательно, все воздействия, которые приводят к сериновому фосфорилированию могут подавлять сигнализацию инсулина. Соответственно, воздействие на клетки-мишени провоспалительных цитокинов (на рис.
7
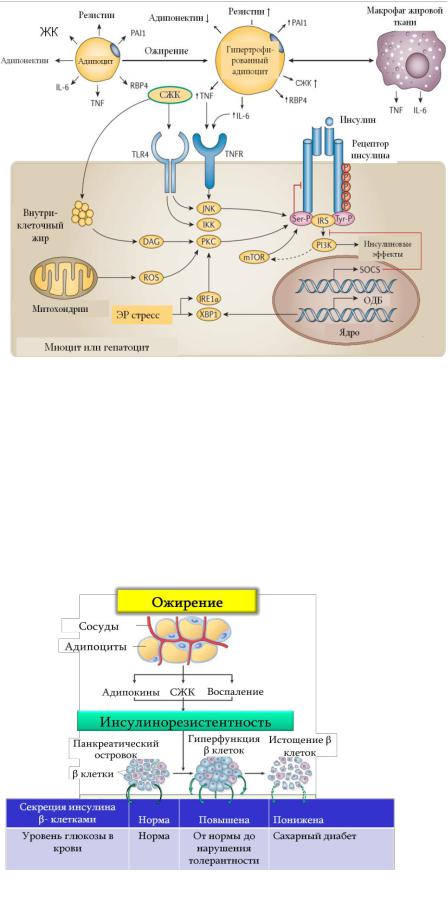
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
12.7 ФНО и ИЛ-6) приводит к сериновому фосфорилированию и, тем самым, к снижению активности инсулиновых рецепторов.
|
|
|
Механизмы |
развития |
ИР наиболее |
|
|
|
хорошо изучены в условиях ожирения. |
||||
|
|
При ожирении, в особенности при |
||||
|
|
абдоминальном |
ожирении, развиваются |
|||
|
|
(рис. 12.8): |
|
|
||
|
|
1) Количественные и качественные из- |
||||
|
|
менения цитокинов - адипокинов, выраба- |
||||
|
|
тываемых жировой тканью: уменьшается |
||||
|
|
продукция адипонектина, |
одновременно |
|||
|
|
увеличивается синтез резистина, ингиби- |
||||
|
|
тора активатора плазминогена (PAI-1), |
||||
|
|
ретинол-связывающего белка (RBP4) и |
||||
|
|
воспалительных цитокинов (рис. 12.7). |
||||
|
|
Провоспалительный «диалог» адипокинов |
||||
|
|
и макрофагов мы подробно рассмотрим в |
||||
|
|
следующей главе. |
|
|
||
Рис. 12.7. Молекулярные механизмы ИР (DeFronzo RA, Ferrannini E, Groop |
2) |
Хроническое |
«вялотекущее» |
|||
L, et al: Type 2 diabetes mellitus, Nat. Rev. Dis. Primers 1:15019, 2015. воспаление, |
|
|
||||
Аббривиатура: TNFR - рецептор ФНО, SOCS (suppressor of |
cytokine |
3) |
увеличение содержания свободных |
|||
singnaling) – ингибитор цитокиновой сигнализации, ОДБ |
- ответ |
|||||
жирных кислот (СЖК) в крови. |
||||||
дефолдированного белков, остальное представлено в тексте. |
|
|||||
|
|
|
|
|
||
|
|
Примечательно, |
что рецепторы TLR-4, |
|||
хорошо знакомые вам по темам «Воспаление» и «Патофизиология иммунной системы», возбуждаются не только их «естественным стимулятором» ЛПС, но также избытком СЖК, подкрепляя воспалительную сигнализацию клетки (JUN - аминоконцевая киназа, киназа IкB – IkB kinase (IKK)) (рис. 12.7 и 12.8).
Вышеизложенные вместе формируют инсулинорезистентность.
Усугублению инсулинорезистентности также способствует митохондриальная дисфункция и стресс эндоплазматического ретикулума (рис. 12.7).
Нарушение функции митохондрий приводит к гиперпродукции свободных радикалов, а стресс ЭР путем активации ответа дефолдированного белка, способствует развитию ряда молекулярных изменений, включая деградацию IRS - субстратов инсулиновых рецепторов.
В начальной стадии островкому аппарату удается поддерживать нормальный уровень глюкозы в крови за счет своей гиперфункции
(гиперинсулинемия) и способствовать проникновению глюкозы в клетки. Однако продолжительное повреждающее действие на β-
клетки дисбаланса адипокинов и ФНО-α, ИЛ-1,
ИЛ-6, СЖК, приводят к их истощению и гибели, и
8
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
постепенно развивается прогресирующая гипергликемия.
Хроническая гипергликемия, способствует десенситизации рецепторов β-клеток, что выражается снижением их секреторной активности. Принимая во внимание патогенное значение хронической гликемии в патогенезе СД, был предложен термин «глюкотоксичность».
Необходимо отметить, что «инкретиновый эффект» значительно снижен у больных тип II СД, и его фармакологическая реактивация приведет к регуляции уровня глюкозы в крови и избыточного веса. Этот принцип привел к созданию двух новых групп препаратов для лечения больных, страдающих тип II СД. Первая группа представляет собой агонисты рецепторов GLP-1, связывающихся и активирующих соответствующие рецепторы β-клеток, а вторая группа включает ингибиторы ферментов DPP-4, под действием которых эндогенные инкретины получают возможность циркулировать и действовать более продолжительно.
Таким образом, патогенез тип II СД может быть представлен в виде следующих событий: первичная ИР и дисфункция β-клеток - действие диабетогенных факторов (стресс, висцеральное ожирение, беременность,
гипокинезия, злоупотребление алкоголем и т.д.) - хроническая гипергликемия - гиперинсулинемия - вторичная ИРнарастающая относительная недостаточность инсулина - истощение -клеток - абсолютная недостаточность инсулина.
Наличие сложных взаимосвязанных патогенетических факторов, участвующих в регуляции углеводного и жирового обменов при тип II СД, позволило G.Reaven в 1988 году сформулировать концепцию метаболического синдрома. Основными компонентами этого синдрома являются ИР тканей и гиперинсулинемия, нарушение толерантности к глюкозе и развитие тип II СД, абдоминальное (висцеральное) ожирение, артериальная гипертензия, дислипидемия и атеросклероз.
Различия между тип I и II диабетами представлены в таблице 12.2.
|
|
|
Таблица 12.2. |
|
|
|
тип I |
тип II |
|
|
|
|
|
|
1.Частота заболеваемости и |
0.2-0.5%, заболеваемость у |
2-4%, женщины болеют чаще. |
|
|
пол |
представителей обоих полов одинакова. |
|
|
|
2. |
Возраст |
Развивается до 20 лет. |
Развивается после 30 лет. |
|
3. |
Наследственная |
У монозиготных близнецов |
У монозиготных близнецов |
|
предрасположенность |
конкордантность 50%. |
конкордантность 90-100%. |
|
|
4. |
Ассоциация с HLA |
HLA ассоциированный. |
Не ассоциированный. |
|
5. |
Циркулирующие антитела |
антитела к инсулину, -клеточной |
Отсутствуют. |
|
|
|
декарбоксилазе глютаминовой кислоты |
|
|
|
|
(GAD), аутоантигену 512 -клеток |
|
|
6. |
Телосложение больных |
худые, низкое содержание жира |
ожирение у 80% |
|
7. |
Секреция инсулина |
прогрессирующее снижение |
в ранней стадии повышенная, а в поздней |
|
|
|
|
стадии нормальная или пониженная |
|
8. |
Морфологические |
инсулит, атрофия Лангергансовых |
в поздних стадиях накопление амилоида в |
|
изменения |
островков |
Лангергансовых островках* |
|
|
9. |
Лечение (основное) |
Иньекции инсулина, без которого |
диета, физическая активность, медикаменты, |
|
|
|
развивается кетоацидоз. |
в конце также инсулин |
|
*При длительном течении тип II диабета в 90% случаев наблюдается накопление амилоида в островках Лангерганса. Некоторые исследователи считают, что амилоид имеет непрямое токсическое воздействие на клетки.
12.1.4. ПРОЯВЛЕНИЯ САХАРНОГО ДИАБЕТА
Проявления сахарного диабета можно разделить на две группы: нарушения обмена веществ и поражения
тканей, органов и систем (последние часто называют осложнениями СД).
9
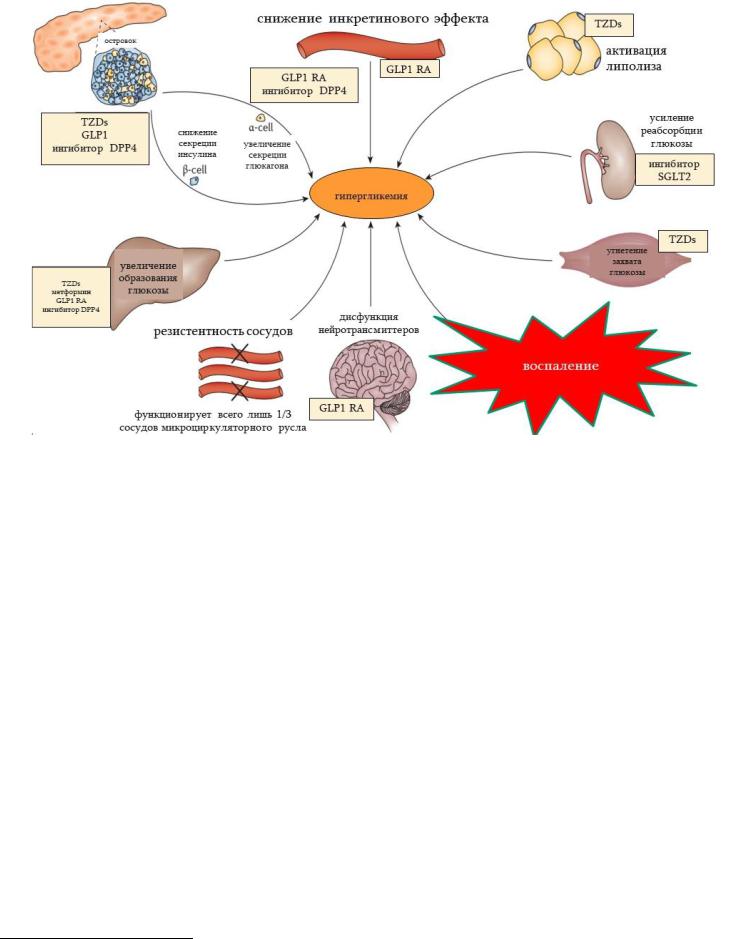
Кафедра Патофизиологии, Патофизиология углеводного обмена, факультет обшей медицины, 2020г.
Рис. 12.9. Механизмы гипергликемии и пути фармакокоррекции. Аббривиатура: GLP1 RA - glucagon-like peptide 1 receptor agonist: агонист рецептора глюкагонаподобного пептида 1; DPP4 inhibitor - dipeptidyl peptidase inhibitor 4: ингибитор дипептидилпептидазы 4; TZDs – thiazolidinediones: тиазолидиндионы (также известные как глитазоны), способствуют усилению потребления глюкозы, а также угнетают липолиз в адипоцитах; SGLT2 - sodium/glucose co-transporter 2: ингибитор Na+/глюкозного котранспортера угнетает реабсорбцию глюкозы в почках.
Проявления нарушений обмена веществ при СД. При СД все обменные процессы нарушаются. Однако ключевым проявлением обменных нарушений при сахарном диабете является гипергликемия.
1. Гипергликемия. Механизмы развития (рис. 12.9):
а) Вследствие недостаточности воздействия инсулина нарушается захват глюкозы клетками (снижение экспрессии GLUT4 транспортеров). В результате позднего осложнения СД - микроангиопатии, количество функционирующих капилляров уменьшается, из-за чего «страдает» прохождение инсулина и глюкозы из крови в ткани (подсчитано, что при тип II диабете фунционирует всего лишь 1/3 микроциркуляторных сосудов).
б) Гликогенолиз усиливается за счет активации гликогенфосфорилазы и глюкозо-6-фосфатазы. Подавляется синтез гликогена (угнетаются глюкокиназа и гликогенсинтетаза). Эти изменения происходят не только из-за дефицита инсулина, но и вследтсвие развивающейся результате этого гиперпродукции глюкагона.
в) Активирован глюконеогенез, поскольку «устранен» репрессивный эффект инсулина (в случае его дефицита)
на ферменты этого метаболического пути.
г) Угнетен процесс преобразования глюкозы в жиры1.
д) В почках усилена реабсорбция глюкозы и в аспекте глюкозурии повышен почечный барьер.
е) По отношению к ряду медиаторов (инсулину, лептину, GLP1, пептиду YY) понижается чувствительность центра насыщения, что приводит к избыточному употреблению пищи – полифагия (встречается при тип II
диабете).
При сахарном диабете гипергликемия имеет двоякое значение:
1 В норме 30% глюкозы под действием инсулина превращается в жиры.
10