

6 курс / Кардиология / Генная_терапия_кардиомиопатий_возможности_и_ближайшие_перспективы
.pdf■ ФУНДАМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ И МЕЖДИСЦИПЛИНАРНЫЕ ТЕХНОЛОГИИ
ДЛЯ КОРРЕСПОНДЕНЦИИ
Лавров Александр Вячеславович – кандидат медицинских наук, ведущий научный сотрудник лаборатории
редактирования генома ФГБНУ «МГНЦ» (Москва, Российская Федерация)
E-mail: avlavrov@yandex.ru https://orcid.org/0000-0003-4962-
6947
Ключевые слова:
неишемические кардиомиопатии; генная терапия; геномное редактирование; РНК-интерференция; антисмысловые олигонуклеотиды; аденоассоциированные вирусные векторы; пропуск экзонов; малые интерферирующие РНК; миРНК
СORRESPONDENCE
Alexander V. Lavrov –
MD, Leading Researcher of the Laboratory of Genome Editing, Research Centre for Medical Genetics (Moscow, Russian Federation) E-mail: avlavrov@yandex.ru https://orcid.org/0000-0003-4962- 6947
Keywords: non-ischemic cardiomyopathies; gene therapy; genome editing; RNA interference; antisense oligonucleotide; adenovirus-associated vectors; exon skipping; small interfering RNA; siRNA
Генная терапия кардиомиопатий:
возможности и ближайшие перспективы
Лавров А.В.1, Заклязьминская Е.В.1–3
1 Федеральное государственное бюджетное научное учреждение «Медико-генетический научный центр имени академика Н.П. Бочкова», 115522, г. Москва, Российская Федерация
2Государственный научный центр Российской Федерации Федеральное государственное бюджетное научное учреждение «Российский научный центр хирургии имени академика Б.В. Петровского», 119991, г. Москва,
Российская Федерация
3Публичное акционерное общество «Центр генетики и репродуктивной медицины “ГЕНЕТИКО”», 119333, г. Москва, Российская Федерация
Первичные кардиомиопатии (КМП) – обширная группа генетически детерминированных заболеваний, проявляющихся функциональным и анатомическим ремоделированием сердечной мышцы в отсутствие очевидных внешних причин. КМП признаны самым частым наследственным заболеванием сердца, их суммарная частота в популяции достигает 0,5%.
Естественное течение всех первичных КМП – неуклонно прогрессирующее, с исходом в сердечную недостаточность. Имеющиеся на сегодняшний день подходы к лечению первичных КМП являются симптоматическими, помогают контролировать симптомы заболевания, но не приводят к полному излечению. Разработка этиологических методов лечения наследственных КМП, которые направлены на коррекцию генетического дефекта, является одной из приоритетных задач современной медицины. В настоящей работе рассмотрены основные разработки в генной терапии заболеваний с дисфункцией миокарда, которые уже появились или близки к выходу в реальную клиническую практику.
Финансирование. Работа выполнена в рамках государственного задания Министерства науки и высшего образования Российской Федерации для ФГБНУ «МГНЦ».
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Для цитирования: Лавров А.В., Заклязьминская Е.В. Генная терапия кардиомиопатий: возможности и ближайшие перспективы // Клиническая и экспериментальная хирургия. Журнал имени академика Б.В. Петровского. 2023. Т. 11, № 1. С. 32–46. DOI: https://doi.org/10.33029/2308-1198-2023-11-1-32-46
Статья поступила в редакцию 09.03.2023. Принята в печать 23.03.2023.
Gene therapy of cardiomyopathies: opportunities and current perspectives
Lavrov A.V.1, Zaklyazminskaya E.V.1–3
1 Research Center for Medical Genetics, 115522, Moscow, Russian Federation
2 Petrovsky National Research Center of Surgery, 119991, Moscow, Russian Federation
3 Centre of Genetics and Reproductive Medicine “GENETICO”, 119333, Moscow, Russian Federation
Non-ischemic (primary) cardiomyopathies (CMP) are a large group of genetically determined diseases manifested by functional and anatomical remodeling of the heart muscle in the absence of obvious external causes. CMP are recognized as the most common hereditary heart disease with a total prevalence 0.5% worldwide.
The natural course of all primary cardiomyopathies is steadily progressive leading to heart failure (HF). Currently available treatment of primary CMP is mainly symptomatic and helps to control various symptoms but does not lead to a complete cure. Etiological approach for the treatment of inherited CMP focused on genetic defect correction, is the priority task of the modern medicine. In this paper, we discuss the main achievements in the gene therapy for primary myocardial dysfunction that have already been introduced or are close to entering the real clinical practice.

Лавров А.В., Заклязьминская Е.В. ■
ГЕННАЯ ТЕРАПИЯ КАРДИОМИОПАТИЙ: ВОЗМОЖНОСТИ И БЛИЖАЙШИЕ ПЕРСПЕКТИВЫ
Funding. The study was financially supported by the State Budget Project of the Ministry of Science and Higher Education of the Russian Federation for Research Centre for Medical Genetics.
Conflict of interest. The authors declare no conflict of interest.
For citation: Lavrov A.V., Zaklyazminskaya E.V. Gene therapy of cardiomyopathies: opportunities and current perspectives. Clinical and Experimental Surgery. Petrovsky Journal. 2023; 11 (1): 32–46. DOI: https://doi.org/10.33029/2308-1198- 2023-11-1-32-46 (in Russian)
Received 09.03.2023. Accepted 23.03.2023.
Первичные кардиомиопатии (КМП) – обширная группа генетически детерминированных заболеваний, проявляющихся функциональным и анатомическим ремоделированием сердеч-
ной мышцы в отсутствие очевидных внешних причин, таких как нарушение коронарного кровотока, перегрузка давлением, врожденные пороки сердца, метаболические расстройства и т.д. [1, 2]. КМП широко распространены во всех этнических группах и в настоящее время признаны самым частым наследственным заболеванием сердца, их суммарная частота в популяции достигает 0,5% [3]. Миокардиальная дисфункция проявляется нарушениями сократимости миокарда, фиброзом сердечной мышцы, нарушениями сердечного ритма и развитием прогрессирующей сердечной недостаточности [3, 4]. Все виды КМП могут манифестировать в любом возрасте. Выявляемость ранних форм КМП составляет примерно 1 случай на 47–50 тыс. детей до 18 лет [5], но чаще диагноз ставится взрослым пациентам.
Естественное течение всех первичных КМП – неуклонно прогрессирующее, с исходом в сердечную недостаточность. Имеющаяся на сегодняшний день лекарственная терапия первичных КМП является симптоматической, помогает контролировать симптомы заболевания, но не приводит к полному излечению. Радикальные хирургические методы лечения [расширенная миоэктомия и алкогольная абляция при гипертрофической кардиомиопатии (ГКМП), обратное ремоделирование при дилатационной кардиомиопатии (ДКМП)] сопряжены с высоким риском интра- и постоперационных осложнений. У многих пациентов существенно улучшаются самочувствие и качество жизни на годы вперед. К сожалению, у некоторых пациентов даже успешное хирургическое лечение является лишь этапом на пути развития тяжелой сердечной недостаточности и необходимости ортотопической трансплантации сердца (ОТС). Учитывая колоссальный разрыв между потребностью в ОТС и возможностью выполнения таких операций, разработка этиологических методов лечения наследственных КМП, которые направлены на коррекцию генетического дефекта, является одной из приоритетных задач современной медицины. В настоящей работе мы
обобщили имеющиеся разработки в генотерапии КМП, которые уже появились или близки к выходу в реальную клиническую практику.
Разнообразие генетических причин кардиомиопатий
В соответствии с доминирующими анатомическими и функциональными изменениями сердечной мышцы, выделяют ДКМП, ГКМП, рестриктивную (РКМП) и аритмогенную кардиомиопатию (АКМП) [6, 7]. Кроме того, синдромные и метаболические КМП, фиброэластоз миокарда, некомпактный миокард (иногда выделяемый в отдельный вариант КМП) объединяют в сборную группу неуточненных (неклассифицированных) КМП [3–7].
Каждая из основных КМП (ГКМП, ДКМП, РКМП и АКМП) также представлена клинически гетерогенной группой заболеваний, которые объединены особым анатомическим и функциональным характером ремоделирования. Исключение составляет только ДКМП, которая, согласно гипотезе общего конечного патогенетического пути [8], может быть как самостоятельным заболеванием, так и исходом практически любой другой КМП. Несмотря на очевидные различия, с генетической точки зрения КМП имеют общие черты. Это преимущественно моногенные заболевания с аутосомно-доминант- ным типом наследования (хотя в 10–15% случаев встречается аутосомно-рецессивное и сцепленное
сполом наследование); доля аутосомно-рецес- сивных и дигенных форм составляет 4–9% [9–12]
сбольшей выраженностью клинической картины заболевания у носителей >1 мутации [12]; огромное генетическое разнообразие (полиаллельность); преимущественно уникальные мутации; значительный внутрисемейный полиморфизм клинических проявлений одного и того же патогенного генетического варианта [1, 4, 7].
Благодаря широкому внедрению методов NGSсеквенирования (NGS, next generation sequencing, секвенирование следующего поколения) спектр генов, ассоциированных с развитием этой группы заболеваний, продолжает расширяться. В общей сложности идентифицировано более 100 генов, патогенные варианты в которых приводят к развитию
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/
ФУНДАМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ И МЕЖДИСЦИПЛИНАРНЫЕ ТЕХНОЛОГИИ
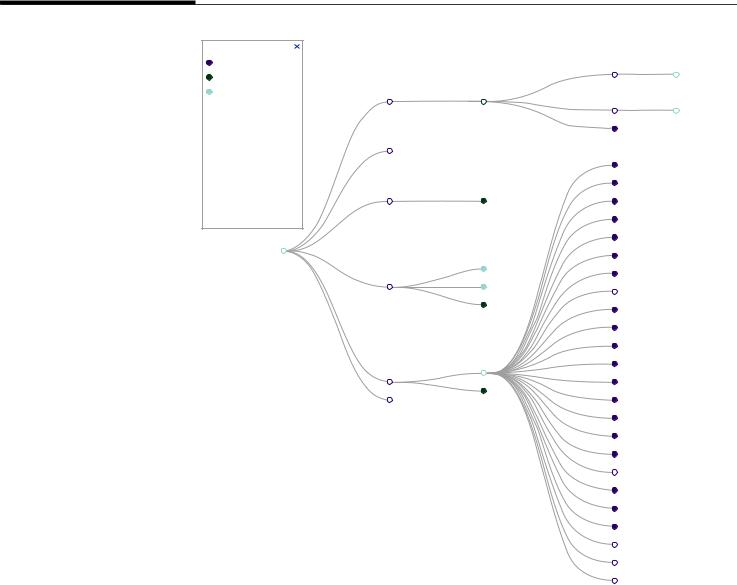
Рис. 1. Взаимосвязь фенотипической серии рестриктивной кардиомиопатии (PS115210)
ссоответствующими генами и другими
фенотипическими
сериями.
Виллюстративных целях раскрыта только часть
связей. Полная картина доступна на сайте OMIM: http://omim.org/graph/ linear/PS115210
Fig. 1. Relationship of the phenotypic series of restrictive cardiomyopathy (RCM, PS115210) with causative genes and other phenotypic series. For clarity, only
partial network is shown. The whole network is available at the OMIM website: http://omim.org/ graph/linear/PS115210
Key: |
|
|
|
|
Phenotype |
|
|
611880: ?Cardiomyopat...(TNN1I3) |
PS115200: Dilated cardiomyop... |
Gene |
|
|
|
|
|
|
|
|
|
Phenotypic series |
115210: Cardiomyopath...(TNNI3) |
191044: TNNI3 |
|
PS192600: Cardiomyopathy, fa... |
(PS) |
|
|
613690: Cardiomyopath...(TNNI3) |
|
• Click circles to show/ |
|
|
613286: Cardiomyopath...(TNNI3) |
|
hide downstream |
|
|
|
|
|
|
|
|
|
relationships |
609578: Cardiomyopath, fa... |
|
|
|
• Click MIM number |
|
|
115195: Cardiomyopath...(TNNT2) |
|
or PS number |
|
|
|
|
|
|
|
|
|
to recenter map |
|
|
115196: Cardiomyopathy...(TPM1) |
|
• Click phenotype |
612422: Cardiomyopath...(TNNT2) |
191045: TNNI2 |
115197: Cardiomyopath...(MYBPC3) |
|
name or gene |
|
|||
|
|
|
||
|
|
|
|
|
symbol to go |
|
|
192600: Cardiomyopath...(CAV3) |
|
to the MIM entry |
|
|
|
|
|
|
|
|
|
PS115210: Familiail restricti... |
|
|
600858: Cardiomyopath...(PRKAG2) |
|
|
|
|
|
|
|
|
|
601493: Left ventricul...(LDB3) |
|
|
PS115200: Dilated cardiomyop... |
607487: Cardiomyopathy...(TCAP) |
|
|
|
|
|
|
|
|
PS192600: Cardiomyopathy, fa... |
|
|
|
615248: Cardiomyopathy...(MYPN) |
|
|
608751: Cardiomyopath...(MYL3) |
|
|
|
608517: MYPN |
|
|
|
|
608758: Cardiomyopathy...(MYL2) |
|
|
|
|
|
|
|
|
|
|
612098: Cardiomyopath...(ACTC1) |
|
|
|
|
612124: Cardiomyopath...(CSRP3) |
|
|
PS192600: Cardiomyopathy, fa... |
612158: Cardiomyopath...(ACTN2) |
|
|
|
|
|
||
617047: Cardiomyopathy...(FLNC) |
|
613243: Cardiomyopath...(TNNC1) |
|
|
102565: FLNC
613251: Cardiomyopathy...(MYH6)
619433: ?Cardiomyopa...(KIF20A) |
613255: Cardiomyopathy...(VCL) |
|
|
|
613690: Cardiomyopath...(TNNI3) |
|
613765: Cardiomyopathy...(TTN) |
|
613838: Cardiomyopath...(MYOZ2) |
|
613873: Cardiomyopath...(JPH2) |
|
613874: Cardiomyopath...(PLN) |
|
613876: Cardiomyopath...(NEXN) |
|
614676: Cardiomyopath... |
|
618052: Cardiomyopath...(ALPK3) |
|
619402: Cardiomyopath...(FHOD3) |
дисфункции миокарда, и этот список еще далеко не полон [1, 4, 6]. Интересно отметить, что многие из этих генов могут вносить свой вклад в развитие всех известных видов КМП, и разные мутации в одном и том же гене реализуются различными вариантами ремоделирования миокарда с формированием так называемых аллельных (фенотипических) серий (рис. 1).
Для многих недавно описанных генетических форм описаны лишь единичные клинические случаи, и патогенетическая роль выявленных редких вариантов все еще требует детального изучения. Поскольку результаты генетического исследования имеют высокое диагностическое значение, в том числе для бессимптомных членов семьи, возникла необходимость оценить доказательность и целесообразность включения все новых генов в существующие диагностические протоколы. Рабочая группа по кардиомиопатиям консорциума ClinGen (https://clinicalgenome.org/affiliation/40008/)
опубликовала серию рекомендаций, в которых был оценен уровень доказательности причинной связи (каузативности) ряда генов в отношении разных типов КМП [13–15], что нашло свое отражение в последних рекомендациях по ДНКдиагностике [2].
Гипертрофическая кардиомиопатия (ГКМП) является наиболее частым типом КМП (1:500– 1:200) [2, 3], и ее генетические причины изучены наиболее полно. Исходно под ГКМП понимали генетически детерминированную гипертрофию миокарда левого желудочка (ЛЖ), вызванную мутациями в генах, кодирующих саркомерные белки миокарда [9]. К настоящему времени описаны мутации более чем в 40 генах, ассоциированных с изолированной ГКМП (OMIM, omim.org; дата обращения 11.02.2023, PS192600), которые относятся как к саркомерным, так и к структурным белкам миокарда, и их регуляторам. Гипертрофия миокарда ЛЖ также является частью сложного симптомоком-

Лавров А.В., Заклязьминская Е.В. ■
ГЕННАЯ ТЕРАПИЯ КАРДИОМИОПАТИЙ: ВОЗМОЖНОСТИ И БЛИЖАЙШИЕ ПЕРСПЕКТИВЫ
плекса различных наследственных заболеваний, таких как нервно-мышечные заболевания (атаксия Фридрейха, дистальная миопатия Лэнга, миотоническая дистрофия и др.), наследственные болезни обмена (болезни Данона, Фабри, TTR-амилоидоз, болезнь Помпе, PRKAG2-связанный гликогеноз
идр.), другие наследственные синдромы (синдромы Нунан, LEOPARD, Костелло, Беквитта–Виде- мана и др.) [15].
Дилатационная кардиомиопатия (ДКМП) также встречается с частотой около 1:500 и является самой частой причиной сердечной недостаточности, требующей ОТС. Не менее половины случаев ДКМП наследственно обусловленные [1–3]. Это заболевание имеет наибольшее число идентифицированных генетических причин в группе КМП. Согласно базе данных менделирующих заболеваний человека (OMIM, omim.org; дата обращения 11.02.2022), к настоящему времени описаны около 100 генов, ассоциированных с развитием ДКМП (PS115200), которые кодируют компоненты сократительного аппарата, ядерной оболочки, клеточной мембраны, ионных каналов, регуляторов метаболизма кальция, регуляторов воспаления, транскрипционных факторов и т.д. [14]. Фенокопии заболевания включают целый ряд прогрессирующих нервно-мышечных (миодистрофии Дюшенна–Беккера, Эмери–Дрейфуса и др.), метаболических заболеваний (болезнь Кернса–Сейра
ит.д.) и синдромных заболеваний (PPP1R13L- обусловленная ДКМП) [16].
Аритмогенная кардиомиопатия правого желудочка (АКПЖ) – термин, которым исторически описывался специфический тип КМП, связанный с мутациями в генах десмосомных белков и характеризующийся очаговым фиброзножировым замещением миокарда преимущественно правого желудочка (ПЖ) и правожелудочковыми аритмиями высоких градаций [17]. Распространенность этого типа КМП оценивается в 1:2500–
1:1000 с более высокой представленностью
внекоторых регионах Италии [18]. Последующие клинические, генетические и патоморфологические исследования показали, что примерно
вполовине случаев пациенты имеют бивентрикулярную форму заболевания, а список заинтересованных генов был расширен до нескольких десятков [19, 20] (фенотипическая серия PS107970 по OMIM, omim.org; дата обращения 11.02.2022). Эти данные послужили толчком для принятия более широкого термина – аритмогенная кардиомиопатия, АКМП. Однако, согласно оценке эксперт-
ной группы консорциума ClinGen, только для 8 генов патогенность в отношении этого заболевания имеет установленный (DSC2, DSG2, DSP, PKP2, JUP и TMEM43) или средний (DES, PLN) уровень доказательности [13].
Рестриктивная кардиомиопатия (РКМП) – более редкий вид КМП, который характеризуется преимущественно диастолической дисфункцией ЛЖ и/или ПЖ (нарушением диастолического наполнения) при относительно сохранной толщине стенок камер сердца и фракции выброса [2]. В среднем РКМП составляет 5–12% всех КМП и характеризуется очень тяжелым прогнозом во всех возрастных группах [21]. Спектр генетических причин РКМП (фенотипическая серия PS115210 по OMIM, omim.org; дата обращения 11.02.2022) неуклонно расширяется, но уже понятно, что наблюдается высокая степень перекрывания с другими группами КМП и других системных наследственных заболеваний (амилоидоз, лизосомные болезни накопления, миофибриллярные миопатии и т.д.) (рис. 1) [2, 6, 21].
Исходом прогрессирования всех типов КМП является сердечная недостаточность (СН), которая в настоящее время является главной причиной смерти от сердечно-сосудистых заболеваний. Не только конкретный вид КМП, но и СН развивается как самостоятельный патологический процесс со своими закономерностями и необратимым ухудшением при отсутствии лечения [21]. При прогрессирующей циркуляторной недостаточности запускаются компенсаторные процессы, связанные с активацией определенных генов, влиять на которые возможно с помощью генной терапии.
Генетическое разнообразие КМП определяет разнообразие разрабатываемых подходов к их лечению. При разработке этиотропного лечения КМП все известные генотерапевтические подходы находят свое применение.
Генная терапия
Генная терапия в начале своего развития подразумевала использование нормальной копии гена вместо мутантной, т.е. представляла собой генозаместительную терапию. Впервые успешно генную терапию применили для лечения дефицита аденозиндезаминазы еще в начале 1990-х годов. Это была генно-клеточная терапия, клетки пациентов модифицировали вирусом ex vivo и вводили обратно. Эффективность лечения была низкой, так как модифицированные клетки плохо приживались, а у части пациентов из модифицированных пересаженных клеток развился лейкоз. Первые успехи генной терапии были окончательно омрачены в 1999 г. смертью пациента с дефицитом орнитинтранскарбамилазы в результате тяжелой иммунной реакции на введенный вирусный вектор с нормальной копией гена. Это событие затормозило прогресс в данной области на долгие годы, а настороженность по отношению к вирусным векторам сохраняется и поныне. Основное беспокой-
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/

ФУНДАМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ И МЕЖДИСЦИПЛИНАРНЫЕ ТЕХНОЛОГИИ
ство касается иммунного ответа и инсерционного мутагенеза, даже несмотря на значительный успех вирусных вакцин от COVID-19 и давно отсутствующую у современных вирусных векторов инсерционную способность.
Фактически второе рождение генозаместительная терапия получила с появлением в 2017 г. одного из самых дорогих препаратов на планете для лечения пигментного ретинита (врожденного амавроза Лебера) – Люкстурна [22]. Действующее вещество препарата – нормальная последовательность гена RPE65, упакованная в аденоассоциированный вирусный вектор. Управление по контролю качества продуктов, лекарственных средств и косметической продукции США (FDA) дало мощный толчок для развития всех направлений генной терапии, в том числе разработкам, направленным на коррекцию генных дефектов на транскрипционном и трансляционном уровнях. В то же время разработки по классической генной терапии были фактически приостановлены, большое внимание стали уделять попыткам использования более простых препаратов, доставка которых не требует использования сложных векторов. Так появились лекарства для пропуска стоп-кодонов и даже целых экзонов, что позволяет восстанавливать функцию транскрипта при ряде патогенных вариантов генов.
Развитие геномного редактирования ознаменовало собой расцвет генной терапии. С момента открытия геномных редакторов в 2012 г. прошло чуть больше 10 лет, а на основе нуклеаз CRISPR/ Cas9 уже зарегистрировано более 80 клинических исследований [23]. Все три подхода в генной терапии используют для разработки препаратов для лечения КМП. Далее будут рассмотрены все генотерапевтические средства, уже зарегистрированные как лекарственные препараты или проходящие клинические исследования фазы III, которые могут быть применены для лечения первичных КМП изолированно и в составе наследственных синдромов.
Генозаместительная генная терапия
Идея внесения в клетку полноценно работающего гена взамен «дефектного» лежит на поверхности, и попытки введения генов с терапевтической целью предпринимали начиная с 1960-х годов. Доставка генной конструкции в клетки живого организма оказалась одним из самых сложных этапов разработки и до сих пор является важным фактором успешности терапии. Самыми эффективными и безопасными в отношении инсерционного мутагенеза и иммуногенности являются последние поколения аденоассоциированных вирусов (ААВ). В последние годы на их основе создано несколько вакцин от коронавируса
SARS-CoV-2, показавших свою безопасность и эффективность у миллионов людей. Единственный существенный недостаток данных векторов – малая пакующая способность, т.е. ограничение на размер вносимого гена, который может нести векторная частица. Тем не менее именно с ААВ на сегодняшний день проводится большинство доклинических
иклинических исследований по генной терапии.
Кдостоинствам ААВ относятся эффективность, длительность персистенции в ядре в эписомном состоянии без интеграции, эффективная экспрессия целевого гена и низкая иммуногенность [24]. ААВ применяют для генной терапии
в многочисленных клинических исследованиях в онкологии, офтальмологии, миологии, неврологии, гематологии и других областях [25–27]. К январю 2023 г. были зарегистрированы 3 препарата для генной терапии на базе ААВ. Первый генотерапевтический препарат (Glybera) получил статус зарегистрированного лекарства в октябре 2012 г. В его составе ААВ нес нормальную копию гена липопротеинлипазы (LPL) для лечения редкой формы семейной дислипидемии [28]. Несмотря на успешную терапию более 30 пациентов, препарат сняли с производства в 2018 г. из-за редкости заболевания и чрезвычайно высокой стоимости препарата.
В последние годы ААВ завоевали огромную популярность, и препараты с их использованием показывают эффективность в десятках клинических и доклинических исследований. Эти конструкции считаются безопасными как с точки зрения вероятности интеграции в геном, так и с иммунологической точки зрения. Определенное беспокойство вызывают только результаты 10-летнего наблюдения за собаками с гемофилией, леченными генной терапией на основе ААВ [29]. Исследователи проанализировали геномы 6 собак и обнаружили 1741 уникальных мест в их геномах, в которых были отмечены вставки генотерапевтической конструкции. Несмотря на потенциальную опасность таких вставок, авторы отмечают, что никаких клинических проявлений вставок в данном исследовании выявлено не было, а клинический эффект генной терапии был продемонстрирован [29]. Важным параметром оценки безопасности во всех исследованиях с использованием вирусных векторов является оценка персистенции как вирусных частиц, так и трансгена, и его возможной интеграции в геном. В целом потенциальный риск современных ААВ, модифицированных таким образом, чтобы максимально снизить интеграционный потенциал, считается низким по сравнению с преимуществами их использования [30]. ААВ могут длительно персистировать в клетке в эписомном состоянии, т.е. в виде ДНК, неинтегрированной в геном, что позволяет обеспечивать длительный

Лавров А.В., Заклязьминская Е.В. ■
ГЕННАЯ ТЕРАПИЯ КАРДИОМИОПАТИЙ: ВОЗМОЖНОСТИ И БЛИЖАЙШИЕ ПЕРСПЕКТИВЫ
терапевтический эффект в неделящихся или медленно делящихся клетках, к которым относятся кардиомиоциты.
Генотерапия миодистрофии Дюшенна
Миодистрофия Дюшенна (МДД, MIM*300377) является одной из самых изученных прогрессирующих мышечных дистрофий. Заболевание развивается в результате потери функции гена дистрофина (DMD) и наследуется по Х-сцепленному рецессивному типу. Частичная потеря функции приводит к развитию более мягкой формы – миодистрофии Беккера. Прогрессирование ДКМП является важным фактором, определяющим прогноз и продолжительность жизни. Усилия десятков научных коллективов и компаний направлены на разработку генной терапии МДД.
SRP-9001
Препарат SRP-9001 (синоним RG 6356, активное вещество delandistrogene moxeparvovec) разработан компанией Sarepta Therapeutics и госпиталем Nationwide Children’s Hospital (США) для генной терапии миодистрофии Дюшенна. Основа препарата – микродистрофин, уменьшенный ген дистрофина, находящийся под промотором, специфичным для мышечной ткани, – MHCK7. Для адресной доставки гена используется генетически модифицированный ААВ серотипа rh47 с усиленной тропностью к мышечной ткани – rAAVrh74. Таким образом, еще одно название препарата – rAAVrh74.MHCK7.microdystrophin. Сейчас препарат SRP-9001 проходит фазу III клинических исследований [clinicaltrials. gov, EMBARK (NCT05096221) – рандомизированное двойное слепое плацебо-контролируемое].
Висследование вошли 126 мальчиков в возрасте от 4 до 7 лет с МДД, вызванной мутациями сдвига рамки считывания или преждевременными стопкодонами между экзонами 18 и 79 (включительно), кроме мутаций в экзоне 45. Среди критериев включения – отсутствие антител к ААВ rAAVrh74.
Впервом плече пациенты получают однократную внутривенную инфузию SRP-9001 и через год – плацебо, а во втором плече наоборот. Оценка клинического эффекта планируется по специализированным шкалам и опросникам, а также по уровню экспрессии микродистрофина через 52 нед после инфузии. Первые результаты планируется анонсировать в 2023 г.
Между тем продолжаются другие более ранние клинические исследования фаз I и II [NCT03375164, NCT03769116, ENDEAVOR (NCT04626674)]. В частности, по данным исследования фазы I–II препарата NCT03375164, проведенном на 4 больных, не было отмечено серьезных побочных эффектов [31]. Наиболее частыми осложнениями были тошнота и транзиентное повышение глутамилтрансферазы,
на фоне приема глюкокортикоидов. Через год после приема препарата у всех пациентов была подтверждена экспрессия трансгена. Белок DMD также был определен иммуногистохимически у всех пациентов в 81% мышечных волокон с правильным расположением вдоль сарколеммы. Уровень креатинфосфокиназы (КФК) снизился, и функциональные тесты (NSAA) показали улучшение по сравнению с началом лечения, с сохранением результатов в течение года после однократной инфузии генотерапевтического препарата.
В ноябре 2022 г. FDA приняло заявку на приоритетное рассмотрение препарата со сроком выдачи решения до 29 мая 2023 г. [32]; таким образом, уже в 2023 г. может появиться первый генотерапевтический препарат для лечения большинства пациентов с МДД.
PF-06939926
Препарат PF-06939926 был разработан компанией Pfizer также для генной терапии МДД. Основой лекарства является мини-дистрофин, аналогичный микродистрофину в SRP-9001, упакованный в модифицированный ААВ9. Клинические исследования фазы III стартовали в ноябре 2020 г. [clinicaltrials. gov, CIFFREO (NCT04281485) – рандомизированное двойное слепое плацебо-контролируемое].
Висследование вошли 99 мальчиков от 4 до 7 лет. Критерии включения в исследование аналогичны описаному выше, критерии исключения были дополнены выводом пациентов со специфической локализацией мутаций в гене DMD (экзоны 9–13, 29 и 30 или делеции, затрагивающие экзоны 56–71). Пациенты из группы плацебо имеют возможность получить препарат через год после первой инфузии.
Вфеврале 2022 г. исследование было приостановлено в связи со смертью пациента в более раннем клиническом исследовании (NCT03362502) того же препарата [33], но в апреле 2022 г. FDA разрешило его продолжить, и в настоящее время идет набор пациентов [34]. Первичные результаты планируются к 2024 г., а окончание запланировано на 2029 г. PF-06939926 стал первым препаратом, зарегистрированным в клинических исследованиях для генной терапии МДД. Его эффективность и безопасность были продемонстрированы в исследовании фазы I (NCT03362502), что позволило получить в FDA статус быстрого прохождения (fast track) в 2020 г. и сразу инициировать исследования фазы III.
Вцелом данные об эффективности и безопасности позволяют предполагать, что PF-06939926 также может получить одобрение FDA в ближайшие 1–2 года.
Помимо SRP-9001 и PF-06939926, в клинических исследованиях (фаза I/II) находится сходный по структуре генотерапевтический препарат
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/

ФУНДАМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ И МЕЖДИСЦИПЛИНАРНЫЕ ТЕХНОЛОГИИ
Рис. 2. Схема сдвига |
мРНК |
А-У-Г-А-А-Г-У-У-У-Г-Г-Ц-А-У-А-Г-У-Г |
||||
рамки считывания |
||||||
в результате делеции |
Нормальная |
|
|
|
|
|
последовательность |
|
|
|
|
|
|
одного нуклеотида |
белок |
Met |
Lys |
Phe |
Gly |
Ile Val |
Fig. 2. Schematic |
|
|
|
У |
|
|
representation |
|
|
|
|
|
|
of a frameshift resulting |
мРНК |
А-У-Г-А-А-Г-У-У-Г-Г-Ц-А-У-А-Г-У-Г |
||||
from a single nucleotide |
||||||
deletion |
Делеция урацила привела |
|
|
|
|
|
к сдвигу рамки считывания |
|
|
|
|
|
|
|
белок |
Met |
Lys |
Leu |
Ala |
Stop |
SGT-001 с опубликованными результатами исследований [33]. Во всех проведенных клинических исследованиях отмечены похожие побочные эффекты. Вероятно, они обусловлены сходными мутациями, приводящими к потере иммуногенного участка дистрофина и, как следствие, иммунными реакциями на этот участок в составе мини/микродистрофина трансгена [35]. Уточнение причин развития осложнений позволит стратифицировать группы пациентов по рискам и уточнить показания и противопоказания к генной терапии.
Генотерапия сердечной недостаточности
Результатом прогрессирования любой КМП является СН. Несмотря на то что СН является конечной стадией различных заболеваний, существуют разработки генной терапии, направленные на лечение именно этого грозного исхода [36]. Исследований с применением ААВ насчитывается несколько десятков [37], а число пациентов, принявших участие в клинических исследованиях, исчисляется сотнями [38]. Однако до сих пор нет одобренных препаратов, и даже до клинических исследований фазы III доведен пока лишь один препарат на основе аденовирусного вектора Ad5.hAC6 (NCT03360448). Аденовирусные векторы в настоящее время применяются гораздо реже ААВ в связи с высокой иммуногенностью.
Ad5.hAC6 (RT-100)
Препарат Ad5.hAC6 (RT-100, Renova Therapeutics) представляет собой ген AC6, кодирующий аденилатциклазу 6 в аденовирусном векторе. Белок АС6 катализирует превращение аденозинтрифосфата (АТФ) в циклический аденозинмонофосфат (цАМФ), который в норме синтезируется в ответ на стимуляцию β-рецепторов и необходим для обеспечения нормальной сократительной функции миокарда. Эффективность и безопасность препарата оценивали в клиническом исследовании фазы I–II у пациентов с фракцией выброса (ФВ) ЛЖ <40%. При однократном внутрикоронарном введении Ad5.hAC6 увеличивал ФВ ЛЖ с 30 до 36% через 4 нед после введения, и этот эффект сохранялся около 8 нед [39]. На основании этого было инициировано клиническое исследование
фазы III (NCT03360448) в 2017 г., однако набор пациентов так и не начали, исследование было отложено для уточнения дальнейшей стратегии. Таким образом, данная разработка до сих пор не доведена до клинического применения и ожидает подтверждения в расширенном клиническом исследовании.
Генетическая коррекция на транскрипционном и трансляционном уровнях
Существует большое разнообразие механизмов реализации патогенных вариантов (мутаций) в генах, приводящих к развитию наследственных заболеваний. При многих из них возможно исправить дефект не только на уровне ДНК гена, но и на уровне его транскрипта. Одним из частых механизмов является формирование неправильного транскрипта и далее белка, что приводит к нарушению нормальной жизнедеятельности клетки. В этих случаях целевое разрушение мутантного транскрипта может иметь терапевтический эффект. В клетке существует несколько механизмов целевого разрушения транскриптов, и для их активации достаточно небольших молекул рибонуклеиновой кислоты (РНК), которые относительно легко можно доставить в организм как простые молекулы в составе большинства привычных лекарств. С помощью малых РНК можно вызвать так называемую РНКинтерференцию, результатом которой будет целевое разрушение патологических транскриптов.
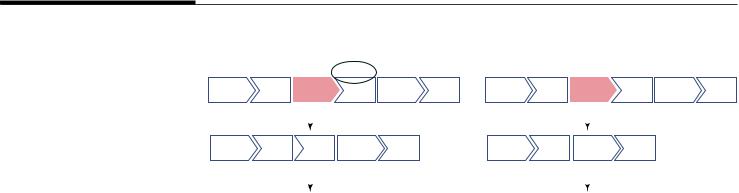
Еще одним механизмом реализации мутаций является образование преждевременного стопкодона либо в результате точковой мутации замены нуклеотидов, либо в результате делеций и вставок, приводящих к сдвигу рамки считывания (рис. 2). В результате заболевание развивается из-за отсутствия или недостаточного количества целевого белка. В этих случаях пропуск стоп-кодонов или даже небольших фрагментов гена (1–2 экзонов) позволяет восстановить экспрессию гена и получить полноценный или несколько укороченный функциональный белок.
Пропуск стоп-кодонов
Преждевременные |
стоп-кодоны возникают |
в результате точковых |
замен нуклеотидов (нон- |

|
|
|
|
|
|
|
Лавров А.В., Заклязьминская Е.В. |
■ |
||
|
|
ГЕННАЯ ТЕРАПИЯ КАРДИОМИОПАТИЙ: ВОЗМОЖНОСТИ И БЛИЖАЙШИЕ ПЕРСПЕКТИВЫ |
|
|||||||
AGG |
T CT |
AAG |
GCA |
A TT |
AAG |
CAG |
AG G |
CAA |
Рис. 3. Схема соединения |
|
50 |
|
51 |
52 |
|
53 |
54 |
|
55 |
экзонов 50–55 гена |
|
|
|
|
дистрофина (DMD) |
|||||||
сенс-мутаций) или в результате вставок/делеций, |
ЕМА выдало условное одобрение для лечения ата- |
||
не кратных 3 нуклеотидам, что приводит к сдвигу |
луреном МДД, вызванной преждевременными стоп- |
||
рамки считывания и возникновению преждевре- |
кодонами, и в 2016 и 2020 гг. пролонгировало его |
||
менного стоп-кодона на расстоянии 20–30 нуклео- |
действие в части рекомендации применения препа- |
||
тидов от места сдвига. Сам по себе такой стоп-кодон |
рата для лечения пациентов с МДД с регулярными |
||
не представляет интереса для таргетной терапии, |
оценками эффективности. |
Данное разрешение |
|
гораздо важнее восстановить рамку считывания, |
действует и сейчас. Из-за низкой эффективности |
||
о чем пойдет речь в следующем разделе. Точковые |
препарата FDA до сих пор не одобрило аталурен |
||
замены, приводящие к появлению преждевремен- |
для лечения МДД, несмотря на то что более 100 па- |
||
ного стоп-кодона, по разным оценкам, являются |
циентов в США получают его в рамках расширен- |
||
причиной моногенных заболевания у 10–15% па- |
ных клинических исследований в течение более |
||
циентов. Например, при МДД преждевременные |
10 лет [46]. По данным метаанализов и некоторых |
||
стоп-кодоны обнаружены у ~13% больных [40]. |
клинических исследований (NCT01557400), аталу- |
||
|
рен как минимум замедляет прогрессию заболева- |
||
Аталурен |
ния [47, 48]. В России препарат был зарегистриро- |
||
В начале 2000-х годов было обнаружено, что |
ван в 2020 г. для лечения пациентов с МДД. |
||
аминогликозиды в высоких концентрациях в клет- |
|
|
|
ках млекопитающих вызывают пропуск преждевре- |
Пропуск экзонов |
|
|
менных стоп-кодонов [41]. В силу многочисленных |
Небольшие делеции и вставки нуклеотидов (ин- |
||
побочных эффектов высоких доз их целевое при- |
делы), обычно 1–2-нуклеотидные, но не кратные |
||
менение для пропуска стоп-кодонов было невоз- |
3-м, являются одними из самых тяжелых точковых |
||
можно. Позднее был проведен скрининг более |
мутаций, так как приводят к сдвигу рамки считыва- |
||
800 тыс. малых молекул и обнаружена одна – |
ния, полному изменению аминокислотной последо- |
||
РТС124, которая эффективно вызывала пропуск |
вательности и появлению стоп-кодонов. Зачастую |
||
стоп-кодонов [42]. РТС124 не затрагивает обыч- |
такие мутации можно скорректировать, если про- |
||
ные стоп-кодоны, так как нуклеотидное окружение |
пустить фрагмент гена (экзон) с ними таким обра- |
||
обеспечивает сборку сложного комплекса терми- |
зом, чтобы рамка считывания восстановилась. Если |
||
нации трансляции [43], который не может столь же |
в область пропуска не попадает никакой функцио- |
||
эффективно собираться вокруг преждевременного |
нальный домен, то сокращенный белок чаще всего |
||
стоп-кодона [44]. РТС124 связывается с трансляци- |
продолжает полноценно функционировать. Неко- |
||
онным комплексом и приводит к тому, что в случае |
торые экзоны в генах стыкуются по границам пол- |
||
преждевременного стоп-кодона рибосома захва- |
ных триплетов (см. рис. 3, экзоны 51–52, 53–54), |
||
тывает транспортную РНК (тРНК) с максимально |
а некоторые с разрывом триплета (50–51, 52–53 |
||
похожим кодом и вставляет соответствующую ей |
и 54–55). Во втором случае протяженная делеция, |
||
аминокислоту, после чего продолжается обычный |
захватывающая целый экзон, сопровождается та- |
||
синтез белка. Белок отличается от нормального |
ким же сдвигом рамки считывания, как и 1–2-нук- |
||
только одной аминокислотной заменой, что в боль- |
леотидные инделы. И в такой ситуации можно вос- |
||
шинстве случаев не сказывается на его функции. |
становить рамку считывания с помощью пропуска |
||
РТС124, названный впоследствие аталуреном, был |
смежного экзона. |
|
|
сразу взят в разработку терапии муковисцидоза |
Идея пропуска экзонов родилась в 1990-е годы, |
||
и МДД. В 2005 г. препарат получил статус орфан- |
и уже через 10 лет появились первые работы по |
||
ного препарата в Европейском медицинском агент- |
пропуску экзонов в гене DMD [49]. В основе дан- |
||
стве (ЕМА). |
ного подхода лежит использование последователь- |
||
В 2017 г. Sarepta Therapeutics прекратила |
ностей, комплементарных |
последовательностям |
|
дальнейшие исследования аталурена как пре- |
РНК гена в области сайтов сплайсинга. Эти по- |
||
парата для лечения муковисцидоза, вызванного |
следовательности называются |
антисмысловыми |
|
преждевременными стоп-кодонами, так как кли- |
олигонуклеотидами (ACO) (рис. 4). Такие олигонук- |
||
ническое исследование фазы III (NCT02139306) |
леотиды препятствуют взаимодействию сплай- |
||
по оценке эффективности аталурена при лечении |
сосомы с РНК, что приводит к вырезанию из зре- |
||
муковисцидоза показало лишь небольшое по |
лой матричной РНК (мРНК) |
смежных экзонов |
|
величине и недостоверное улучшение функции |
в составе прилегающих интронов. Такого же эф- |
||
внешнего дыхания [45]. Однако еще в 2014 г. |
фекта можно добиться, если заблокировать не сами |
||
Fig. 3. Schematic representation of the exons 50–55 junctions in the dystrophin gene (DMD)
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/
ФУНДАМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ И МЕЖДИСЦИПЛИНАРНЫЕ ТЕХНОЛОГИИ
|
|
|
|
|
Сплайсосома |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
и энхансеры |
|
|
|
|
|
|
|
Антисмысловой |
|
|
|
|
|
|
сплайсинга |
|
|
|
|
|
|
|
олигонуклеотид |
|
50 |
51 |
52 |
|
53 |
54 |
55 |
50 |
51 |
52 |
53 |
54 |
55 |
||
|
|
|
Сплайсинг |
|
|
|
|
|
Сплайсинг |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||||
50 |
51 |
|
53 |
54 |
55 |
|
50 |
51 |
54 |
55 |
|
|
||
|
|
|
Нарушение трансляции |
|
|
|
|
Трансляция |
|
|
||||
|
|
|
|
|
|
|
|
|
||||||
Сдвиг рамки считывания и отсутствие дистрофина |
|
|
Восстановление рамки считывания и синтез |
|
||||||||||
|
|
|
|
|
|
|
уменьшенного, но функционирующего дистрофина |
|||||||
Рис. 4. Принцип действия антисмысловых олигонуклеотидов (АСО). При делеции экзона (красным выделен экзон 52) в результате сплайсинга происходит соединение оставшихся экзонов 51 и 52 со сдвигом рамки считывания. В этом случае нормальный белок дистрофин не синтезируется, а полученные транскрипты деградируют. АСО блокирует посадку комплекса сплайсосомы, или энхансеров сплайсинга, распознающих определенные последовательности внутри экзонов, в результате экзон 53 вырезается из зрелого транскрипта в составе интронов, а сплайсинг завершается соединением экзонов 51 и 54, в результате чего рамка считывания вновь восстанавливается, и возможен синтез дистрофина с выпавшим участком белка, кодируемого экзонами 52–53. Такой дистрофин сохраняет значительную часть своей функции
Fig. 4. The principle of action of the antisense oligonucleotides (ASO). Deletion of the exon 52 (highlighted in red) results in the junction of the exons 51 and 52 with a frame-shift. In this case, the normal dystrophin protein is not synthesized, and the resulting transcript undergoes degradation. ASO blocks formation of the spliceosome or splicing enhancers that recognize certain sequences within exons. As a result, exon 53 is skipped from the mature transcript together with introns. The exons 51 and 54 are connected restoring the reading frame. Now dystrophin’s synthesis is possible again with a shortened protein missing a small part encoded by exons 52–53. Such a dystrophin retains a significant part of its function.
сайты сплайсинга, а особые последовательности внутри экзонов, необходимые для их распознавания белками, участвующими в сплайсинге. В современных препаратах используют измененные – морфолиновые олигонуклеотиды, в которых вместо дезоксирибозы в остов пентозофосфатной цепи входят морфолиновые кольца. Такие олигонуклеотиды не вызывают разрушения комплементарных молекул, а только блокируют доступ к соответствующим последовательностям. Кроме того, они более стабильны по сравнению с нативными олигонуклеотидами и лучше проникают в клетки [50]. Пропуск экзонов как терапевтический подход возможен для многих наследственных заболеваний, но лучше всего изучена МДД. В настоящее время FDA зарегистрированы несколько препаратов для лечения МДД, действующих по принципу пропуска экзонов.
Касимерсен (Amondys 45)
Касимерсен (Sarepta Therapeutics) зарегистрирован FDA в 2021 г. для лечения МДД [51], вызванной мутациями, эффект которых может быть компенсирован пропуском экзона 45. Препарат вводят еженедельно внутривенно капельно. По предварительным данным клинических исследований фазы III (NCT02500381), касимерсен хорошо переносится и обеспечивает увеличение уровня дистрофина с 0,93 до 1,74% нормы, что, по мнению FDA, должно положительно сказаться на течении заболевания [52].
Этеплирсен( Exondys 51)
Этеплирсен (Sarepta Therapeutics) является первым препаратом для пропуска экзонов, получившим одобрение FDA в 2016 г. [53]. Совокупные данные 7 клинических исследований, включая 1 исследование фазы III (NCT02255552), позволяют утверждать, что этеплирсен не только увеличивает экспрессию нормально расположенного под сарколеммой дистрофина, но и положительно влияет на клиническую картину заболевания: у части пациентов увеличивается дистанция ходьбы в функциональных тестах, у других заболевание хоть и прогрессирует, но несопоставимо медленнее, чем в группе пациентов, не получавших терапию [53].
Голодирсен (Vyondys 53), Витоларсен (Viltepso)
Голодирсен (Sarepta Therapeutics) зарегистрирован FDA в 2019 г. [54], витоларсен (NS Pharma, дочка Nippon Shinyaku Co.) – в 2020 г. [55] для лечения МДД, вызванной мутациями, эффект которых может быть компенсирован пропуском экзона 53. Голодирсен хорошо переносится и, по данным клинических исследований (NCT02310906), обеспечивает до 0,92% нормального количества дистрофина [54]. Синтезируемый дистрофин имеют правильную локализацию под сарколеммой, а у пациентов показана остановка прогрессии заболевания. Продолжаются клинические исследования фазы III по оценке отдаленных функциональных эффек-

Лавров А.В., Заклязьминская Е.В. ■
ГЕННАЯ ТЕРАПИЯ КАРДИОМИОПАТИЙ: ВОЗМОЖНОСТИ И БЛИЖАЙШИЕ ПЕРСПЕКТИВЫ
тов терапии, прежде всего сохранения или восстановления физической активности (NCT02500381, NCT03532542).
Витоларсен также одобрен по ускоренной процедуре и до сих пор проходит клинические исследования фазы III (NCT04060199). По данным клинических исследований I/II фазы (NCT02740972, NCT03167255 и Japic CTI-163291), препарат очень хорошо переносится и повышает продукцию дистрофина в мышцах через 20–24 нед после начала терапии [56]. К сожалению, пока нет данных о функциональном улучшении у пациентов, принимающих терапию. Есть только отдельные сообщения от представителей компании, проводящей исследования, что прием препарата останавливает функциональное ухудшение, которое наблюдают у пациентов, не получающих данной терапии [57]. Оценка функционального эффекта является главной целью проводимых исследований фазы III. Препарат также вводят еженедельно.
РНК-интерференция
Семейство антисмысловых олигонуклеотидов (АСО) очень разнообразно. Некоторые АСО могут участвовать в так называемой РНК-интер- ференции – одном из вариантов снижения экспрессии гена путем «выключения» аллеля (сайленсинга) на посттранскрипционном уровне. Основа РНК-интерференции – сиквенс-специфичное разрушение транскриптов. При РНК-интерференции короткие молекулы РНК или ДНК связываются с комплементарными им молекулами мРНК и привлекают особые белки, разрушающие мРНК одним из двух основных путей. В случае малых интерферирующих РНК (миРНК) собирается белковый RISC-комплекс, блокирующий или разрушающий транскрипт, что в обоих случая приводит к снижению трансляции белка. В случае антисмысловых одноцепочечных ДНК их дуплекс с мРНК распознается РНКазой H, которая расщепляет связанный транскрипт.
Соответственно их применение возможно при заболеваниях, вызванных появлением белка с нарушенной функцией или конформацией. Снижение или выключение экспрессии мутантного аллеля сохраняет аллель с нормальной функцией, и таким образом достигается терапевтический эффект. Одним из заболеваний, при котором РНК-интерференция показала высокую эффективность, является транстиретиновый амилоидоз, который может скрываться за маской ГКМП.
Транстиретиновый амилоидоз (ТТА) – наследственное аутосомно-доминантное заболевание, вызываемое мутациями в гене транстиретина (TTR). Транстиретин вырабатывается преимущественно в печени, связывает и поддерживает сывороточ-
ную концентрацию гормонов Т3 и Т4 и участвует
вметаболизме витамина А. Мутации гена нарушают сборку белка и приводят к его отложению
вамилоидных фибриллах. Накопление амилоида происходит медленно, и заболевание проявляется
ввозрасте от 20 до 60 лет. Клинические проявления ТТА зависят от положения мутаций в гене. Мутации в начале гена ассоциированы с доми-
нирующей периферической полиневропатией, а расположенные более дистально – к преобладанию накопления амилоида в сердце, гипертрофии миокарда и прогрессирующей СН [58]. Наличие КМП обычно ассоциировано с более тяжелым течением ТТА.
Генная терапия ТТА направлена на снижение экспрессии транстиретина, что уменьшает отложение амилоида и проводится с использованием обоих подходов – АСО и миРНК-интерференции.
Антисмысловые олигонуклеотиды
Антисмысловые олигонуклеотиды (АСО) представляют собой модифицированные одноцепочечные молекулы ДНК, обладающие высокой про- теин-связывающей способностью. В случае ТТА они взаимодействуют с рецепторами на поверхности гепатоцитов, проникают в клетку и дальше в ядро, где обеспечивают разрушение мРНК транстиретина [59]. В настоящее время FDA зарегистрированы два препарата (инотерсен и эплотерсен).
Инотерсен (Akcea Therapeutics) был одобрен FDA в 2018 г. по результатам клинических исследований фазы III (NCT04136184). В исследование вошли 172 пациента, в том числе с развившейся КМП (NYHA1-2). Инотерсен показал положительный эффект в отношении неврологической симптоматики, но для оценки влияния на гипертрофию миокарда данных было недостаточно [60]. Побочные эффекты (тяжелая тромбоцитопения, гломерулонефрит) развились суммарно у 6% пациентов, причем все были носителями одной мутации p.V30M).
Эплонтерсен (АстраЗенека, известен также как AKCEA-TTR-LRx, разработанный компанией Akcea Therapeutics) проходит клинические исследования фазы III. Препарат является полным аналогом инотерсена с той же нуклеотидной последовательностью, но конъюгирован с N-ацетилгалактозамином. По данным разработчиков, это позволяет улучшить доставку молекулы в гепатоциты и ослабить нагрузку купферовских клеток и эндотелиоцитов, в которые попадает препарат [61]. После проникновения в клетки конъюгат отщепляется и высвобождается инотерсен, взаимодействующий с мРНК TTR [62]. Эплонтерсен хорошо переносится, не вызывает серьезных побочных эффектов и, по имеющимся данным, не требует отмены ни у кого из пациентов [63]. При этом эплонтерсен в большей
Рекомендовано к изучению сайтом МедУнивер - https://meduniver.com/