

nature reviews disease primers
Primer
https://doi.org/10.1038/s41572-023-00465-y
 Check for updates
Check for updates
Hirschsprungdisease
Louise Montalva |
1,2,3 , Lily S. Cheng4,5, Raj Kapur 6, Jacob C. Langer7, Dominique Berrebi8, Kristiina Kyrklund9, |
Mikko Pakarinen |
9, Ivo de Blaauw10, Arnaud Bonnard1,2,3 & Ankush Gosain 11 |
Abstract |
Sections |
Hirschsprung disease (HSCR) is a rare congenital intestinal disease |
Introduction |
|
that occurs in 1 in 5,000 live births. HSCR is characterized by the |
|
|
Epidemiology |
||
absence of ganglion cells in the myenteric and submucosal plexuses |
|
|
Mechanisms/pathophysiology |
||
of the intestine. Most patients present during the neonatal period with |
||
|
||
Diagnosis, screening and |
||
the frst meconium passage delayed beyond 24 h, abdominal distension |
||
prevention |
||
and vomiting. Syndromes associated with HSCR include trisomy 21, |
|
|
Management |
||
Mowat–Wilson syndrome, congenital central hypoventilation syndrome, |
||
Quality of life |
||
Shah–Waardenburg syndrome and cartilage–hair hypoplasia. Multiple |
||
|
||
putative genes are involved in familial and isolated HSCR, of which |
Outlook |
|
the most common are the RET proto-oncogene and EDNRB. Diagnosis |
|
|
consists of visualization of a transition zone on contrast enema and |
|
|
confrmation via rectal biopsy. HSCR is typically managed by surgical |
|
|
removal of the aganglionic bowel and reconstruction of the intestinal |
|
|
tract by connecting the normally innervated bowel down to the anus |
|
|
while preserving normal sphincter function. Several procedures, namely |
|
|
Swenson, Soave and Duhamel procedures, can be undertaken and may |
|
|
include a laparoscopically assisted approach. Short-term and long-term |
|
|
comorbidities include persistent obstructive symptoms, enterocolitis |
|
|
and soiling. Continued research and innovation to better understand |
|
|
disease mechanisms holds promise for developing novel techniques |
|
|
for diagnosis and therapy, and improving outcomes in patients. |
|
1Department of Paediatric Surgery, Robert-Debré Children’s University Hospital, Paris, France. 2Faculty of Health, Paris-Cité University, Paris, France. 3NeuroDiderot, INSERM UMR1141, Paris, France. 4Division of Paediatric Surgery, Texas Children’s Hospital, Houston, TX, USA. 5Division of Paediatric Surgery, University of Virginia, Charlottesville, VA, USA. 6Department of Pathology, Seattle Children’s Hospital, Seattle, WA, USA. 7Division of Paediatric Surgery, The Hospital for Sick Children, Toronto, Ontario, Canada. 8Department of Pathology, Robert-Debré
and Necker Children’s University Hospital, Paris, France. 9Department of Paediatric Surgery, Helsinki University Central Hospital, Helsinki, Finland. 10Department of Surgery, Division of Paediatric Surgery, Radboudumc-Amalia Children’s Hospital, Nijmegen, Netherlands. 11Department of Paediatric Surgery, Children’s Hospital Colorado, Aurora, CO, USA.  e-mail: louise.montalva@aphp.fr; ankush.gosain@childrenscolorado.org
e-mail: louise.montalva@aphp.fr; ankush.gosain@childrenscolorado.org
Nature Reviews Disease Primers | |
(2023) 9:54 |
1 |

Primer
Introduction
Hirschsprung disease (HSCR) is a rare congenital intestinal disease characterized by the absence of ganglion cells in the distal rectum, extending for variable distances into the proximal intestine. This lack of ganglion cells causes a functional blockage in the intestine, leading to symptoms such as constipation, abdominal distension and vomiting. Approximately 80% of patients with HSCR are diagnosed during the neonatal period (during the first 4 weeks of life), with the first passage of meconium frequently delayed beyond 24 h (ref. 1).
Based on the length of the aganglionic segment of the intestine, HSCR is classified into two types — short-segment HSCR and long-segment HSCR. Although the exact definition of these terms can vary between studies, the current standardized nomenclature defines long-segment HSCR as extension of the aganglionic bowel beyond the sigmoid or descending colon junction2 (Fig. 1). HSCR is a broad-spectrum disease, as some patients can be affected by aganglionosis of the whole colon (total colonic aganglionosis) or even the whole intestine (total intestinal aganglionosis), resulting in <20 cm of normally innervated bowel, which is associated with high mortality3,4. HSCR occurs as a result of incomplete migration of enteric neural crest cells (NCCs) during embryo development, and is associated with mutations in several genes such as RET, GDNF, EDNRB, SOX10 and PHOX2B leading to defective signalling pathways5.
HSCR is suspected upon clinical and radiological signs of intestinal obstruction or severe chronic constipation, but formal diagnosis requires a rectal biopsy in order to confirm aganglionosis that is often associated with submucosal nerve hypertrophy6. The identification of aganglionosis and abnormal peristalsis as disease aetiology has substantially improved the outlook in patients with HSCR since the 1920s (Fig.2).Initialmanagementrequiresboweldecompression,whichmay beachievedusingrectalwashoutsinshort-segmentHSCRorviaadivert- ing stoma in long-segment HSCR. The definitive treatment of HSCR involvesthesurgicalremovaloftheaganglionicbowel.The‘pull-through’ reconstruction procedure described in 1949 by Orvar Swenson involving the removal of the aganglionic bowel and creating an anastomosis betweenthenormallyinnervatedbowelandtheanalcanal,remainsthe standard surgical approach for HSCR today7. However, as rectal dissectionbylaparotomyininfantsistechnicallydifficultandcanresultinhigh rates of complications, other pull-through techniques were developed and several techniques are still widely used today8–13.
Whereas the initial pull-through procedure required a combined laparotomy and transanal approach, two less invasive approaches were developedinthemid1990sandarenowwidelyusedassingle-stagepro- ceduresinshort-segmentHSCR:thelaparoscopy-assistedpull-through and the pure transanal pull-through14–16. Despite surgical management, children with HSCR can experience both short-term and long-term complications, including ongoing obstructive symptoms, episodes of enterocolitis and issues with faecal continence17,18. Several research questions regarding preoperative planning, the optimal surgical procedure and postoperative care remain to be addressed in order to improve the quality of life of patients with HSCR.
This Primer offers a comprehensive summary of the up-to-date research on HSCR, covering various aspects such as its epidemiology, pathophysiology, diagnosis, management and effect on quality of life, and discusses future directions for research.
Epidemiology
HSCR is the most common congenital motility disorder of the gut19. Worldwide prevalence ranges between 1.0 and 2.6 per 10,000 live births
and varies across the world, potentially owing to true geographical, ethnic and population differences1,20–22. Incidence rates have also been reported,andrangebetween0.3and4.1per10,000livebirths23,24.These differences might also possibly be biased by severe disparities in sample sizes and differences in registry systems1,20–22 (Table 1). Although two registry-based studies have shown a slight increase in the prevalence of HSCR between 1980 and 2005, this observation may be influenced by improved adherence to database capture over time. HSCR is considered a sex-dependent, multifactorial genetic disorder in which environmental factors seem to play a minor part compared with genetic factors, although studies have described several risk factors for HSCR.
Risk factors
Sex distribution. HSCR affects males more frequently than females, with a ratio ranging from 2.8:1 to 4.0:1 (refs. 1,21,25,26). However, this pattern does not hold true in syndromic HSCR (defined as those cases of HSCR associated with other chromosomal disorders), which has a sex ratio of 1:1, or in long-segment HSCR or total colonic aganglionosis, which has a ratio of 1.2:1.9 (refs. 21,27).
Maternal factors. There is limited knowledge regarding maternal factors that can increase the risk of HSCR. One study found that maternal obesity and having more than three children were risk factors for HSCR21. Another study found maternal age >30 years was a risk factor28, although subsequent studies failed to confirm this finding1,21. HSCR is also often associated with genetic disorders, which tend to have an increased prevalence with increasing maternal age.
Ethnicity. Studies have revealed variations in prevalence and the incidence of HSCR according to ethnicity within certain study populations (Table 1). For example, in the USA, African Americans, Pacific Islanders and Asians have a higher incidence of HSCR than Hispanics23,28. These variations suggest that there may be differences in genetic mutation frequencies among different populations, with certain variants being more common in certain populations26,29. For example, in children of South-East Asian ancestry, common and rare variants of NRG1 have been found to be associated with the development of HSCR, whereas these variants seem to be less frequent in the white population30–32. Similarly, variants in RET have been found to differ across ethnicities and could explain some of the global differences observed in HSCR incidence29,33. However, larger studies are needed to better understand these subtle differences and to avoid making broad generalizations based on limited data.
Intestinal segment length. Long-segment HSCR is known to have a dominant inheritance pattern with incomplete penetrance while short-segment HSCR is often transmitted in an autosomal recessive fashion33. The length of the aganglionic segment is thought to be influenced by the severity of mutations, the number of copies of the mutations present and the presence of modifying factors, such as epigenetic and environmental factors.
Preterm infants.As diagnosing HSCR in preterm infants might be challenging for surgeons and pathologists owing to less specific symptoms and ganglionic immaturity, systematic reviews of >4,000 infants found that only6–7% of children with HSCRwere preterm, suggesting that the incidence may be lower among premature babies than among full-term babies34,35. However, the incidence of prematurity among children with HSCR has increased since 2000s, possibly due to increased disease
Nature Reviews Disease Primers | |
(2023) 9:54 |
2 |
Primer
a
Total intestinal HSCR: <1%
• <20 cm of ganglionated intestine beyond the ligament of Treitz
Small intestinal HSCR: 5%
• Aganglionosis extending beyond the terminal ileum (>5 cm)
Total colonic HSCR: 8%
• Aganglionosis of the entire colon and of <5 cm of the terminal ileum
 Long-segment HSCR: 15%
Long-segment HSCR: 15%
•Aganglionosis from sigmoid colon–descending colon junction up to the caecum (ganglion cells present in colon)
Short-segment HSCR: 72%
•Aganglionosis to the sigmoid colon–descending colon junction
b |
c |
d |
e |
f |
g |
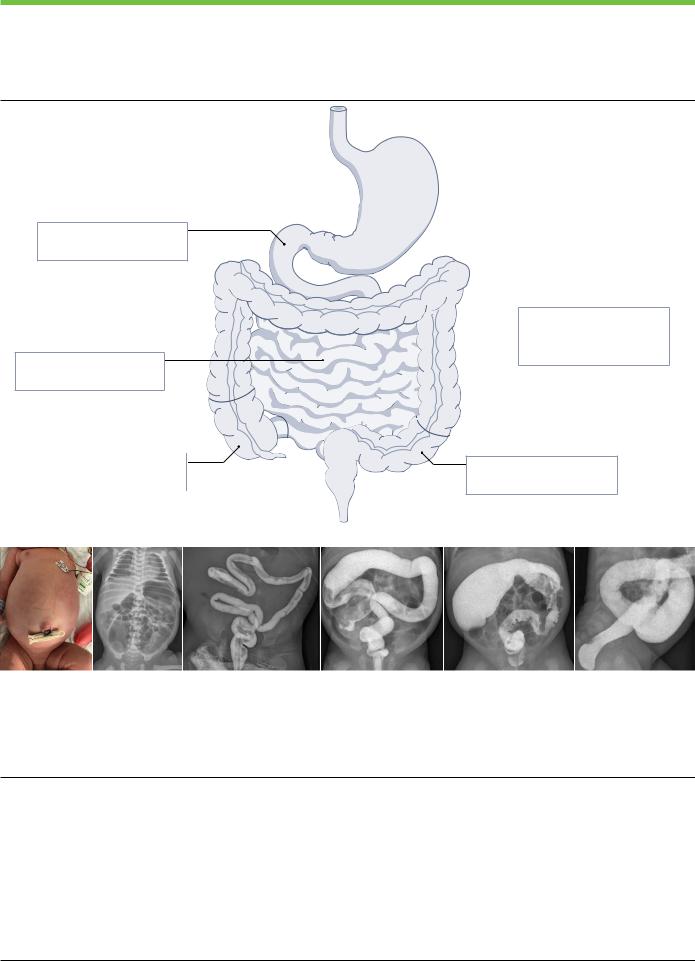
Fig. 1 | Clinical and radiological diagnosis of Hirschsprung disease.
Recommended Hirschsprung disease (HSCR) nomenclature (part a): subtypes and incidence depending on length of aganglionosis2. Abdominal distension in a male neonate with delayed meconium passage and bilious vomiting (part b). Plain abdominal radiograph shows diffuse intestinal distension and little air in the pelvis in a neonate with HSCR (part c). Contrast enema shows a microcolon
in a neonate with small-intestinal HSCR, extending beyond the terminal ileum (part d); a short ‘question-mark’-shaped colon (part e); a transition zone in the transverse colon, with a distended caecum and a narrow distal colon, with irregular saw-tooth contractions (part f); and a transition zone on the sigmoid colon and a rectosigmoid index of <1 (part g).
awareness andalsoduetoan increasedincidence ofprematurity,which is estimated to be ~11% worldwide35,36. However, evidence indicates that the enteric nervous system (ENS) reaches anatomical and functional maturation by mid-gestation in humans, and therefore preterm birth might not affect the incidence of HSCR37.
Associated syndromes. In 70% of children with HSCR, the disease is an isolated condition but it can also be familial, associated with genetic syndromes or chromosomal anomalies33,38. Chromosomal anomalies areobservedin12%ofpeoplewithHSCR,withtrisomy21beingthemost
common1. Down syndrome has an overall incidence of 7.3% in children with HSCR and the incidence of HSCR in children with Down syndrome is 2.6%39. Other syndromes that are frequently associated with HSCR include Waardenburg–Shah syndrome (with pigmentary disorders), Haddad syndrome (with congenital hypoventilation), Mowat–Wilson syndrome and other sporadic syndromes (Table 2). Although additional syndromes continue to be reported as associated with HSCR, owing to the rarity of the disease and its low penetrance, determining whether the association of HSCR with a syndrome is coincidental or a true element of the syndrome is challenging38. Most genetic variations
Nature Reviews Disease Primers | |
(2023) 9:54 |
3 |