

Атлас_редких_болезней_2016_2_е_издание
.pdfУважаемые коллеги!
Перед вами второе издание атласа, посвященного одному из главных направлений современной медицины — редким болезням у детей. Своевременная диагностика, правильное ведение и адекватная реабилитация пациентов с орфанными заболеваниями являются проблемой не только в нашей стране, но и во всем мире. Практически все болезни, относящиеся к данной категории, обусловлены генетически, имеют неуклонно прогрессирующее течение, а значит, приводят к нарушению функции пораженных органов, инвалидизации, потере трудоспособности, навыков самообслуживания, к смерти. Таким образом, все вышеперечисленное возводит редкие болезни в ранг не только медицинских проблем, но и социальных, общечеловеческих, а значит, государственных.
Достижения современной науки (молекулярной биологии, генетики, физиологии, патофизиологии и др.) позволяют выяснить природу многих болезней, которые ранее считались патологией с неясной этиологией и патогенезом. Это привело к созданию этиологически направленной, зачастую заместительной, высокотехнологической терапии, эффективно контролирующей течение многих болезней, ранее считавшихся некурабельными.
Предлагаемый вашему вниманию обновленный «Атлас редких болезней» содержит описание уникального многолетнего опыта, наработанного сотрудниками ведущего педиатрического медицинского учреждения нашей страны — Научного центра здоровья детей. В издании лаконично представлена вся современная информация, необходимая для того, чтобы врач любой специальности, увидев перед собой пациента с редкой патологией, смог провести положенное исследование по месту жительства или направить больного в федеральный центр для точной постановки диагноза и назначения адекватной терапии. Возможности диагностики в нашей стране, в том числе уровень развития молекулярно-гене- тических методов исследования, не уступают развитым зарубежным странам и позволяют нашим маленьким пациентам получать эффективное лечение, не покидая пределов Российской Федерации.
Несомненно, врачи, владеющие знаниями, смогут вовремя распознать среди множества других часто встречающихся заболеваний орфанное, правильно поставить диагноз и назначить своевременное, основанное на принципах доказательности лечение, а значит, сохранить жизнь еще одному ребенку.
Министр здравоохранения |
В.И. Скворцова |
Российской Федерации |
|
10
ПРЕДИСЛОВИЕ
С первого января 2012 г. вступил в силу Федеральный закон № 323-ФЗ «Об охране здоровья граждан Российской Федерации», в котором впервые дано понятие редких (орфанных) болезней. К этой категории болезней относят заболевания, распространенность которых не превышает 10 случаев на 100 тысяч населения, и для лечения которых требуется дорогостоящее медикаментозное сопровождение. Именно поэтому пациенты с орфанными заболеваниями представляют собой социально наименее защищенную группу населения. Включив в законодательство понятие орфанных болезней, Российская Федерация сделала первый шаг к успешному решению проблемы обеспечения необходимым лечением всех своих граждан.
В перечень редких (орфанных) заболеваний, подготовленных Минздравом России, входят наследственные болезни обмена веществ, крови и кроветворных тканей, а также ряд наследственных моногенных синдромов, для которых разработано патогенетическое лечение.
К сожалению, врачи общей практики и педиатры недостаточно хорошо осведомлены о клинических проявлениях и методах диагностики большинства орфанных болезней, а в части случаев подтвердить заболевание не представляется возможным в связи с удаленностью от крупных федеральных центров.
Вместе с тем необходимо отметить, что успехи последних лет в автоматизации и повышении чувствительности аналитических инструментов, а также применение новых методов исследования (например, тандемная масс-спектрометрия) значительно упрощает и ускоряет выявление врожденных нарушений обмена веществ и других орфанных заболеваний. Новые достижения в диагностике и лечении наследственных болезней приводят к тому, что многие из них не только успешно лечатся, но и рассматриваются как кандидаты для неонатального скрининга. Предложены скринирующие тесты для лизосомных болезней накопления, миодистрофии Дюшенна, болезни Вильсона и др. Новые технологии, такие как мультиплексный анализ белков и пептидов, являются одним из способов упрощения и ускорения лабораторной диагностики. Это с успехом продемонстрировано для лизосомных болезней накопления и может быть применено к более широкому кругу заболеваний. За последние несколько лет достижения, связанные с расшифровкой генома, ускорили открытие индивидуальных вариантов генов, носительство которых непосредственно указывает на связь с тем или иным заболеванием. Все это способствовало разработке и внедрению в медицинскую генетику значительного числа новых молекулярно-генетических методов и технологий, дающих возможность выявления заболеваний, которые нельзя обнаружить с помощью традиционных биохимических методов анализа. Так, технология, использующая микрочипы для одновременного исследования десятков и сотен полиморфных участков в различных областях генома, позволяет выявлять значимые мутации, существенно сокращая при этом время анализа. Другое перспективное направление технологий на основе микрочипов — это быстрое определение уровня и профиля экспрессии сотен и тысяч генов, специфически экспрессирующихся в данном органе или ткани, с выявлением тех
11

Атлас редких болезней
генов, экспрессия которых нарушена или отсутствует по причине генетических нарушений (мутаций). В ряде исследований уже показаны впечатляющие возможности полногеномного сканирования на образцах сухих пятен крови, и даже сделаны предположения о возможности полного геномного секвенирования у новорожденных с использованием малоинвазивных методов забора биологического материала.
Редкие (орфанные) заболевания в большинстве своем относятся к группе заболеваний с прогредиентным течением и ранним летальным исходом. Именно поэтому ранняя диагностика дает возможность своевременно подобрать пациенту эффективную терапию. Неонатальный скрининг данной группы заболеваний, основанный на современных методах молекулярно-генетической диагностики, может служить тем краеугольным камнем, который поможет своевременно установить диагноз, определить прогноз и даже спасти множество жизней.
Безусловно, использование разработанных методов нуждается в широком обсуждении и осторожном принятии решений в связи с возникающими этическими проблемами, связанными с выявлением носительства патологических генов или досимптоматическим обнаружением болезней, не имеющих эффективных методов лечения. Однако, темпы развития науки существенно опережают описание новых нозологических форм, и, следовательно, социальную значимость раннего выявления таких болезней невозможно переоценить.
Мы надеемся, что эта книга поможет ознакомиться широкому кругу педиатров
иврачей других специальностей с клиническими проявлениями редких (орфанных) болезней для раннего выявления, своевременного и адекватного лечения
ипрофилактики возникающих осложнений.
Директор Научного центра здоровья детей, Главный внештатный специалист педиатр Министерства здравоохранения РФ, Председатель Исполкома Союза педиатров России, академик РАН
А.А. Баранов
Директор НИИ педиатрии Научного центра здоровья детей — заместитель директора Центра по науке, заведующая кафедрой аллергологии и клинической иммунологии
педиатрического факультета Первого МГМУ им. И. М. Сеченова, заведующая кафедрой факультетской педиатрии № 1 педиатрического факультета РНИМУ им. Н. И. Пирогова, член Исполкома Международной педиатрической ассоциации (IPA),
президент Европейской педиатрической ассоциации EPA/UNEPSA, советник ВОЗ, член-корреспондент РАН, профессор
Л.С. Намазова-Баранова
12

РАЗДЕЛ I.
РЕДКИЕ
НАСЛЕДСТВЕННЫЕ
БОЛЕЗНИ
АБЕТАЛИПОПРОТЕИНЕМИЯ
(ABETALIPOPROTEINAEMIA)
МКБ-10: Е78; ОМIМ 200100
Определение. Абеталипопротеинемия — редкое врожденное заболевание с мультисистемными проявлениями, характеризующееся нарушением всасывания жиров, акантоцитозом и гипохолестеринемией.
Сгодами у пациентов прогрессирует дефицит жирорастворимых витаминов,
втом числе с формированием атипичного пигментного ретинита, «ночной слепоты», коагулопатии, нейропатии и миопатии. Впервые заболевание описано F.A. Bassen и A.L. Kornzweig в 1950 г.
Синонимы: синдром Бассена–Корнцвейга.
Эпидемиология. Распространенность меньше 1: 1000000 населения.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. Причина абеталипопротеинемии — абсенс-мута- ция функциональной единицы 97-KDa гена МТТР (Microsomal Triglyceride Transporter Protein), расположенного на коротком плече 4-й хромосомы (4q22-24), ответственного за экспрессию белка — микросомального транспортера триглицеридов. Ген, ответственный за продукцию-липопротеинов, в частности аполипопротеина В (хиломикронов), приводит к нарушению синтеза и всасывания триглицеридов, которые необходимы для всасывания жирорастворимых витаминов (А, D, Е, К) в тонкой кишке и транспорта их в печень. Нарушение синтеза липопротеидных комплексов приводит к дефекту клеточных мембран, который наиболее очевиден при исследовании эритроцитов, приобретающих форму звездчатых акантоцитов.
Клинические проявления
В период новорожденности и грудном возрасте — тяжелая мальабсорбция жиров, гипохолестеринемия с рождения; младенцы выживают при своевременной диагностике или получающие полное парентеральное питание.
Непереносимость жиров сохраняется всю жизнь, на мальабсорбцию жира не влияет применение высоких доз липазы.
Хронический дефицит липопротеидов сказывается на структуре мембран всех клеток организма. В частности нарушается структура реснитчатого эпителия, что приводит к дефекту мукоцилиарного клиренса (обструктивным бронхитам, трахеиту, хроническим инфекциям ротоносоглотки), отмечаются эрозивные дефекты слизистой оболочки верхнего отдела пищеварительного тракта (кислотозависимого генеза, репарация слизистой оболочки страдает).
Пигментная ретинопатия.
14

Абеталипопротеинемия
Диагностика
Клиническая симптоматика: нарушения всасывания в кишечнике, а также (вследствие демиелинизации аксонов) нарушения координации, атаксия, нистагм, пигментная дегенерация сетчатки, стеатогепатит, отставание в физическом и умственном развитии.
Лабораторная диагностика: гипохолестеринемия, дефицит липопротеидов низкой и очень низкой плотности; коагулопатия (обусловлена дефицитом витамина К), акантоциты в мазке крови.
Акантоциты периферической крови: 60–85% в мазках, макроцитарная анемия (MCV 97,4 fl). На фоне лечения: акантоцитов 16%, MCV 80 fl
Дифференциальный диагноз:
•муковисцидоз;
•аутосомно-рецессивный нейроакантоцитоз и др. (табл.)
Таблица. Дифференциально-диагностический алгоритм симптомов абеталипопротеинемии
Симптом |
Особенности |
Дифференциальный диагноз |
|
|
|
|
|
|
|
Эхиноциты — эритроциты, поверхность которых |
|
|
|
равномерно покрыта треугольными выростами, |
|
|
|
могут появиться при уремии. |
|
|
Шпоровидные |
Акантоциты (с 5–10 шипообразными |
|
|
эритроциты |
отростками) обнаруживают при аутосомно- |
|
Акантоциты |
рецессивных нейроакантоцитозах (акантоциты |
||
(эхино- |
|||
|
и прогрессирующая дегенерация базальных |
||
|
и акантоциты) |
||
|
|
ганглиев), хореоакантоцитозе и Х-сцепленном |
|
|
|
синдроме McLeod, абеталипопротеинемии. |
|
|
|
При этих заболеваниях акантоциты необходимо |
|
|
|
отличать от эхиноцитов |
|
|
|
|
|
|
|
Муковисцидоз, врожденная гипоплазия |
|
|
|
поджелудочной железы (синдромы Швахмана– |
|
Мальабсорбция |
Первичная |
Даймонда, Йоханссона–Близзарда, Пирсона и др.), |
|
врожденная недостаточность панкреатической |
|||
(стеаторея) |
(врожденная) |
||
липазы и колипазы, врожденная атрезия желчных |
|||
|
|
||
|
|
ходов, дефицит энтерокиназы, врожденная |
|
|
|
атрофия/гипотрофия тонкой кишки |
|
|
|
|
15

Атлас редких болезней
Симптом |
Особенности |
Дифференциальный диагноз |
|
|
|
|
|
|
|
Кахексия, голодание; синдром мальабсорбции; |
|
|
|
обширные ожоги; тяжелые острые заболевания |
|
|
|
и инфекции; некроз гепатоцитов, терминальная |
|
|
|
стадия цирроза печени, гепатокарцинома; |
|
|
|
сепсис; гипертиреоз; хроническая сердечная |
|
|
|
недостаточность; гипо- и абеталипопротеинемия, |
|
|
|
клинически сходные заболевания с разной |
|
|
|
генетической основой (АPO-B мутацией |
|
Гипохолес- |
С рождения |
с доминантным наследованием при семейной |
|
теринемия |
гипобеталипопротеинемии и MTTP при |
||
|
|||
|
|
абетапротеинемии); дефицит -липопротеина |
|
|
|
(болезнь Танжера); мегалобластическая анемия; |
|
|
|
талассемия; хронические обструктивные |
|
|
|
заболевания легких, туберкулез легких; прием |
|
|
|
препаратов, снижающих уровень холестерина; |
|
|
|
прием некоторых лекарственных препаратов |
|
|
|
(кломифена, эстрогенов, интерферона, |
|
|
|
неомицина, тироксина, кетоконазола) |
|
|
|
|
|
|
Снижение |
Аутосомно-рецессивная форма ПР, |
|
|
остроты |
аутосомно-доминантная форма с медленным |
|
|
ночного |
прогрессированием: Х-сцепленная рецессивная — |
|
Пигментная |
зрения |
наиболее тяжелая форма пигментного ретинита |
|
ретинопатия |
(следствие |
с полной потерей зрения на 4-м десятилетии |
|
(ПР) |
дефицита |
жизни. ПР при синдроме Рефсума, |
|
|
витамина А) |
липофусцинозе, мукополисахаридозах типов I, |
|
|
и сужение |
II и III, синдроме Барде–Бидля, наследственной |
|
|
полей зрения |
атаксии и миотонической дистрофии |
|
|
|
|
Лечение. Заключается в назначении высоких доз жирорастворимых витаминов, и особенно токоферола.
Прогноз заболевания зависит от формирования цирроза печени вследствие стеатогепатита, отложения эфиров холестерина, развития атаксии (обычно 3-я декада жизни) и слепоты к 4-й декаде жизни вследствие пигментной дегенерации сетчатки. Дефицит кератинов, витамина А приводит к гиперплазии слизистой оболочки желудка, двенадцатиперстной кишки.
Клинический пример
Мальчик Г., возраст 10 лет 8 мес (рис.).
Наследственный анамнез: кровнородственный брак, 3-я беременность (1-я — здоровая девочка; 2-я — ребенок умер в месячном возрасте, предположительно от кишечной инфекции: отмечалась аналогичная клиническая картина полифекалии, приведшая к эксикозу; девочка от 4-й беременности здорова).
16
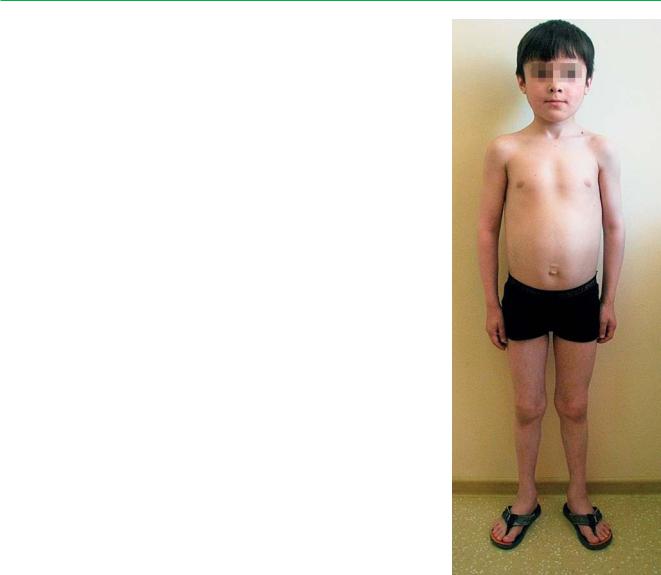
Абеталипопротеинемия
Анамнез жизни: вес и рост при рождении в нор- |
|
|
ме; с месячного возраста перестал прибавлять в весе, |
|
|
полифекалия и стеаторея при хорошем аппетите. |
|
|
За первый год жизни неоднократно госпитализи- |
|
|
ровался с целью парентерального питания, после |
|
|
1 года жизни отмечалось относительное улучше- |
|
|
ние на низкожировой диете. Многократно исклю- |
|
|
чался муковисцидоз. После формирования цирроза |
|
|
и портальной гипертензии, морфологически под- |
|
|
твержденных, направлен в гастроэнтерологическое |
|
|
отделение НЦЗД. |
|
|
При поступлении: вес ребенка 22 кг, рост 117 см |
|
|
(< 3 , отставание 12–20 см), состояние по заболева- |
|
|
нию средней тяжести, самочувствие удовлетвори- |
|
|
тельное. Внешний вид: кукольный, инфантильный. |
|
|
Весо-ростовые показатели и пропорции тела соот- |
|
|
ветствуют 7-летнему возрасту. На лице — сосудистые |
|
|
звездочки, капиллярит. Из носа гнойные выделения. |
|
|
Гнойный конъюнктивит, блефарит. Продуктивный |
|
|
кашель, проводные хрипы. Гепатомегалия +4–5 см, |
|
|
спленомегалия +3 см. |
|
|
Результаты обследования |
|
|
Установлены относительная и абсолютная лим- |
|
|
фопения — 11% (норма 20–50%) и 1,0 109/л |
|
|
(1,0 109/л); макроцитарная анемия с повышени- |
|
|
ем среднего объема эритроцитов — CMV 97,4 fl |
|
|
(77–94 fl), 60–85% акантоцитов; дефицит вита- |
|
|
мина В12 — 177 пг/мл (208–964); повышенная |
Внешний вид ребенка 10 лет |
|
активность креатинфосфокиназы — 361 (25–194) |
8 мес с абеталипопротеинемией, |
|
и лактатдегидрогеназы 290 МЕ/л (91–225); гипохо- |
статус соответствует 6–7 г. |
|
лестеринемия 1,2 ммоль/л (3,1–5,2 ммоль/л); АЛТ |
Костный возраст |
|
от календарного не отстает |
||
44 ед/л, АСТ 93 ед/л (5–40); гепатит минимальной |
||
|
||
степени активности, гамма-глютамилтранспептида- |
|
за (ГГТП) в норме. Низкое содержание витамина В12 в сочетании с мегалобластной анемией стало основанием для назначения цианокобаламина. Эластография печени 5,9 кПа соответствует фиброзу 1-й степени (по шкале Metavir), диагностическая точность 88,6%, ложноотрицательные результаты могут быть связаны с жировой инфильтрацией печени, характерной для абеталипопротеинемиии. Костный возраст соответствует календарному.
Установлен этиологический диагноз абеталипопротеинемии на основании специфического липидного статуса и акантоцитоза, отражающего дефект мембран эритроцитов.
Проведено лечение: витамин Е по 9 капсул в день (100 мг/кг в сут) + витамин А по 1 капсуле в день, альфакальцидол по 0,25 мкг 2 раза в день, менадион (Викасол) по 15 мг 2 раза в день, витамин В12 по 1,0 в/м № 4, препарат карнитина (Элькар 20% в дозе 10 мл/сут), комплекс витаминов и метаболитов (Цитофлавин
17

Атлас редких болезней
по 1/2 таблетки 2 раза в день), урсодезоксихолевая кислота (Урсофальк по 250 мг на ночь); местная терапия конъюнктивита. Амбулаторно рекомендовано продолжить ежедневный прием витаминов Е и А, гепатопротектора.
За 8 мес амбулаторного наблюдения вырос на 10 см, перестал болеть респираторными инфекциями, улучшился профиль коагуляции: протромбиновый индекс (ПТИ) увеличился с 43 до 73% (норма 70–100%).
Список рекомендованной литературы
1.Bassen F.A., Kornzweig A.L. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood. 1950; 5: 381–387.
2.Berriot-Varoqueaux N., Aggerbeck L., Samson-Bouma M., Wetterau J.R. The role of the microsomal triglyceride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000; 20: 663–697.
3.Jung H., Danek A., Walker R. Neuroacantocytosis syndromes. Orphanet J Rare Dis. 2011; 6: 68–69.
4.Shils M.E., Olson J.A., Shike M. Modern nutrition in health and disease. Lea & Febiger. 1994. URL: http://www.vitamini.ru
5.Rader D., Brewer H. Abetalipoproteinemia. New insights into lipoprotein assembly and vitamin E metabolism from a rare genetic disease. JAMA. 1993; 270: 865–869.
6.Singh V. Low LDL Cholesterol (Hypobetalipoproteinemia) updated Jan 3. 2012. URL: http://emedicine. Medscape.com/article/121975-overview
7.Zamel R., Khan R., Pollex R., Hengele R. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis. 2008; 3: 19–29.
18

АКРОДЕРМАТИТ ЭНТЕРОПАТИЧЕСКИЙ
(ACRODERMATITIS ENTEROPATHICA)
МКБ-10: E83.2; ОМIМ 201100
Определение. Акродерматит энтеропатический — наследственный дерматоз, обусловленный мальабсорбцией цинка в тонком кишечнике в результате врожденного отсутствия фермента олигопептидазы; характеризуется везикулобуллезными высыпаниями (преимущественно вокруг естественных отверстий и на дистальных отделах конечностей), алопецией и диареей.
Синонимы: синдром Данбольта–Клосса, синдром Брандта.
Эпидемиология. Распространенность в популяции составляет 1:5000–1:6500. Чаще встречается среди детей, рожденных от близкородственного брака.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. В основе болезни лежит мутация гена SLC39A4, расположенного на хромосоме 8 q24.3, который кодирует трансмембранный белок — цинк-связывающий фактор (ЦСФ). Развивается нарушение процессов всасывания в кишечнике за счет дефицита цинка и отсутствия фермента олигопептидазы (гидролизует олигопептиды, возникающие при распаде белков). У здоровых лиц всасывание цинка осуществляется в двенадцатиперстной кишке с помощью цинк-
|
А |
Б |
Пациент Р., 7 месяцев. Диагноз: «Энтеропатический |
А. Пациент Р., 5 лет (наблюдение в динамике), |
|
акродерматит». В течение 2 месяцев лечился |
проявления энтеропатического акродерматита |
|
в стационаре районной больницы с диагнозом: |
(глоссит и перианальный дерматит с кандидозной |
|
«Атопический дерматит» без клинического |
инфекцией) на фоне прекращения приема препарата |
|
эффекта |
цинка в течение месяца из-за его отсутствия |
|
|
в аптечной сети |
|
Б. Пациент Р., 5 лет 3 месяца (наблюдение в динамике), регресс клинических проявлений на фоне возобновления приема препарата цинка. В настоящее время мальчику 12 лет: при ежедневном приеме препарата цинка обострений кожного процесса не отмечается
19