

UMKA_11651_35_03_07-02_30_08_17OP_58773
.pdfСопоставляя спектры предполагаемых компонентов пробы, выясняют возможность определения одних веществ в присутствии других. Если спектры компонентов пробы накладываются друг на друга, результаты анализа смеси будут завышенными. Для снижения систематических погрешностей, связанных с наложением спектров, созданы
особые приемы измерений и расчета результатов. Другие выходы из положения - маскирование или предварительное отделение мешающих компонентов. Аппаратура. Для
измерения оптической плотности растворов и регистрации спектров поглощения используют спектрофотометры (рис.3).Важнейшая их часть - монохроматор. Другие узлы - источник света, приемник излучения и регистрирующее устройство. Источники света. В зависимости от оптической области, в которой работает прибор, источниками света служат: в УФ-области – водородная или дейтериевая газоразрядные лампы, дающие сплошной спектр излучения; в видимой области – обычная лампа накаливания с вольфрамовой нитью, в ИК-области –глобар. Это керамический стержень, нагреваемый до температур порядка 1600 0С. Монохроматоры. В спектрофотометрах применяют призменные монохроматоры или дифракционные решетки. Материал, из которого изготавливают оптическую систему прибора, должен хорошо пропускать свет в рабочем диапазоне длин волн. В УФ-области используют кварц, в видимой области –стекло, в ИКобласти – кристаллические соли, галогениды щелочных и щелочноземельных металлов
(NaCl, KBr, CaF2 ).
Спектр поглощения — зависимость показателя поглощения вещества от длины волны (или частоты, волнового числа, энергии кванта и т. п.) излучения. Он связан с
энергетическими переходами в веществе. Для различных веществ спектры поглощения различны[1].
Исторически первые наблюдения линейчатых оптических спектров поглощения в спектре Солнца проделал в 1802 году Волластон, но не придал открытию значения, поэтому эти линии были названы «фраунгоферовыми» в честь другого учёного Фраунгофера, который детально изучил их в 1814—1815 гг
Измерения спектров поглощения могут проводиться как с источником белого света, так и с источниками монохроматического излучения.
Для почти свободных атомов и молекул в разрежённых газах, оптический спектр поглощения состоит из отдельных спектральных линий и называется линейчатым.
Разным веществам соответствуют разные спектры поглощения, что позволяет использовать спектроскопические методы для определения состава вещества. Для твёрдых веществ спектры поглощения непрерывны, но встречаются и отдельные линии.
Спектр поглощения F-центров в кристалле NaCl
Спектр поглощения — зависимость показателя поглощения вещества от длины волны (или частоты, волнового)
1. 5 Лекция №5( 2 часа).
Тема «Электрохимические методы анализа Потенциометрия»
1.5.1Вопросы лекции:
1.Электрохимические методы анализа
2.Потенциометрия.
1.5.2Краткое содержание вопросов
Электрохимические методы анализа (ЭХМА) основаны на использовании процессов, которые протекают на поверхности электродов или в приэлектродном пространстве.
Аналитическим сигналом в ЭХМА может служить любой электрический параметр, связанный с составом раствора и концентрацией вещества в нём, например, потенциал (Е), сила тока (I), сопротивление (R), электрическая проводимость (W), количество электричества (Q).
Названия конкретных методов чаще всего связаны с измеряемыми электрическими параметрами: потенциометрия, амперометрия,кондуктометрия и т. д.
Основные узлы приборов электрохимических методов анализа Приборы электрохимических методов анализа, несмотря на всё их многообразие, содержат одни и те же основные узлы: электрохимическую ячейку, устройство для измерения электрического параметра и внешние металлические проводники.
Гальванические элементы используются в потенциометрии, кондуктометрические ячейки, в которых электроды выполняют одинаковую функцию, – в кондуктометрии. В методах, основанных на протекании электролиза, применяются электролитические ячейки.
В качестве устройств для измерения электрических параметров служат микроамперметры (измерение силы тока I), милливольтметры (измерение разности потенциалов Е), мосты переменного тока (измерение сопротивления R), кондуктометры (измерение электрической проводимости W) и др. Внешние металлические проводники осуществляют связь электрохимической ячейки с устройством для измерения электрического параметра. Потенциометрия
Потенциометрией называют группу методов количественного анализа, основанных на использовании зависимости равновесного потенциала электрода, опущенного в
раствор, от активности (концентрации) ионов этого раствора.
Теоретической основой потенциометрии является уравнение Нернста. Практически потенциометрия реализуется путем создания на основе исследуемой системы гальванического элемента.
В потенциометрии используют гальванические элементы, в которых потенциал одного из
электродов зависит от концентрации исследуемого вещества. Этот электрод называют рабочим (индикаторным) или электродом измерения.Второй электрод в потенциометрии являетсяэлектродом сравнения.Этот электрод выбирают с таким
расчетом, чтобы его потенциал оставался постоянным и не зависел от состояния исследуемого раствора.
В таком случае измеренная э.д.с. будет зависеть только от потенциала индикаторного электрода. На основании изменения величины э.д.с. можно судить о концентрации вещества в исследуемом растворе.
Потенциометрические измерения являются надежными при изучении констант равновесия электродных реакций, коэффициентов активности ионов в растворе, констант нестойкости комплексных ионов, рН растворов.
Потенциометрические методы анализа имеют ряд преимуществ. Они особо чувствительны и не требуют для исследования больших объемов растворов. Существуют модификации потенциометрического определения, позволяющие проводить анализ в пробах, объем которых может не превышать десятых долей миллилитра, что важно для
биологических исследований. Поскольку равновесное значение потенциала устанавливается быстро, то потенциометрические измерения не требуют значительных затрат времени. Их можно проводить в мутных и окрашенных растворах, вязких средах.
Различают прямую и косвенную потенциометрию или потенциометрическое титрование.
Прямая потенциометрия (ионометрия)– это потенциометрический метод, в котором индикаторным электродом является ионоселективный электрод. Ионометрия – удобный, простой, экспрессный современный метод анализа. Для его реализации достаточно подобрать соответствующий ионоселективный электрод для определяемого иона. Особенно широко ионометрия используется при определении точной концентрации ионов Н+в растворе. В качестве индикаторного электрода, как правило, применяют стеклянный электрод, селективный по отношению к ионам Н+. Измерения э.д.с. производят с помощью специальных приборов, называемых рН-метрами. Их шкала откалибрована таким образом, что вместо измеряемой э.д.с. показывает значение рН раствора.
Потенциометрическое титрованиея вляется разновидностью титриметрических методов анализа. При потенциометрическом титровании анализируемый раствор, находящийся в электрохимической ячейке, титруют подходящим титрантом, фиксируя точку эквивалентности на основании характера изменения э.д.с. измеряемой цепи в зависимости от объема добавляемого раствора. По полученным данным строят кривую потенциометрического титрования и на этой кривой определяют точку эквивалентности и объем израсходованного титранта в точке эквивалентности.
Потенциометрическое титрование может быть использовано для определения концентрации кислоты или основания в анализируемом растворе. В этом случае составляют гальваническую цепь, содержащую индикаторный стеклянный электрод и электрод сравнения с постоянным известным потенциалом (например, хлорсеребряный):
Э.д.с. такой гальванической цепи будет зависеть от концентрации ионов Н+в исследуемом растворе. Измеряя её с помощью соответствующего прибора, можно непосредственно определить рН среды.
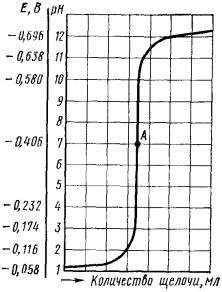
Титруемый раствор строго определенного объема помещают в электролитическую ячейку с электродами и при перемешивании из бюретки небольшими порциями добавляют титрант (например, раствор щелочи при определении концентрации кислоты в исследуемой системе). После каждого добавления очередной порции раствора щелочи с помощью потенциометра определяют рН раствора и затем по полученным данным строят кривую титрования в координатах рН раствора – V(титранта)
Рис. Кривая потенциометрического титрования соляной кислоты гидроксидом натрия: А – точка эквивалентности
Вслучае титрования сильной кислоты щелочью точка эквивалентности будет лежать на середине скачка титрования (рН = 7).
Вряде случаев чувствительность потенциометрического метода превышает чувствительность обычного объемного метода, а также метода кондуктометрического титрования
1. 6 Лекция №6( 2 часа). Тема Вольтамперметрия
1.6.1Вопросы лекции:
1.Вольтамперметрия
1.7.2 Краткое содержание вопросов
Вольтамперометрическими называют методы анализа, основанные на регистрации и изучении зависимости тока, протекающего через электролитическую ячейку, от внешнего наложенного напряжения. Графическое изображение этой зависимости называется вольтамперограммой Для регистрации вольтамперограмм используют индикаторный электрод и электрод
сравнения. В зависимости от типа индикаторного электрода вольтамперометрические Методы принято делить полярографию и собственно вольтамперометрию. Вольтамперометрия – один из электрохимических методов анализа, основанный На использовании явления поляризации микроэлектрода, получении И интерпретацииполяризационных кривых, отражающих зависимость силы тока от приложенного напряжения. В вольтамперометрии используют два электрода: рабочий поляризуемый электрод с малой поверхностью и неполяризуемый электрод сравнения.
При прохождении постоянного тока через электролитическую ячейку процесс характеризуется соотношением:
E = Eа – Eк + I · R,
где E – приложенное извне напряжение; Eа – потенциал анода; Eк –потенциал катода; I – ток в цепи; R – сопротивление электролитической ячейки.
2 При вольтамперометрических измерениях анализируемый растворсодержит индифферентный электролит большой концентрации, поэтому R =1 кОм, а ток не превышает 10-5А, отсюда падением напряжения на ячейке пренебрегают.Если потенциал рабочего электрода измерять относительно потенциала электрода сравнения, условно приняв последний за нуль, то E = Eа для рабочего микроэлектрода и E = - Eк для рабочего микрокатода. Таким образом регистрируемая вольтамперограмма отражает электрохимический процесс, происходящий только на одном электроде. Если в растворе присутствуют вещества, способные электрохимически восстанавливаться или окисляться, то при наложении на ячейку линейно изменяющегося напряжения кривая ток-потенциал имеет форму волны (вольтамперограмма, или полярограмма При низких значениях потенциала на рабочем микроэлектроде не протекает электрохимическая реакция При увеличении потенциала электрохимически активное вещество вступает в электрохимическую реакцию на электроде и ток резко возрастает (рис. 1, В).
При дальнейшем росте потенциала он достигает некоторое предельное значение, далее оставаясь постоянным . Если в качестве рабочего выбран электрод с постоянно обновляющейся поверхностью (например, ртутный капающий электрод), То метод анализа называют полярографическим.
3 В основе качественного анализа лежит величина потенциала полуволны (Е1/2), характеризующая природу деполяризатора: чем менее отрицателен потенциал, тем легче протекает восстановление. Количественный анализ основан на прямой зависимости диффузионноготока I (иливы соты волны h) от концентрации анализируемого вещества:
I = k · c ,
где k – постоянная, зависящая от природы электрода и определяемого
вещества; c – концентрация исследуемого вещества, моль/ дм3. Способы количественного определения концентрации вещества:
1)метод градуировочного графика;
2)метод стандартов;
3)метод добавок.
1. 7 Лекция №7 ( 2 часа).
Тема Хроматографические методы и их классификация
1.7.1Вопросы лекции:
1.Хроматографические методы и их классификация
1.7.2 Краткое содержание вопросов Хроматография –это физико-химический метод разделения веществ, основанный
на распределении компонентов между двумя фазами – подвижной и неподвижной. Неподвижной фазой обычно служит твердое вещество (сорбент) или пленка жидкости, нанесенная на твердое вещество. Подвижная фаза представляет собой жидкость или газ, протекающий через неподвижную фазу.
Компоненты анализируемой смеси вместе с подвижной фазой перемещаются вдоль стационарной фазы, которую обычно помещают в колонку (стеклянную или металлическую трубку). Если молекулы разных компонентов разделяемой смеси обладают различной адсорбируемостью или растворимостью, то время их пребывания в неподвижной фазе, а следовательно, и средняя скорость передвижения по колонке различны. Одни компоненты остаются в верхнем слое сорбента, другие, с меньшей адсорбируемостью, оказываются в нижней части колонки, некоторые покидают колонку вместе с подвижной фазой. Так достигается разделение компонентов. Хроматография – динамический метод, связанный с многократным повторением сорбционных и десорбционных процессов, так как разделение происходит в потоке подвижной фазы. Это обеспечивает эффективность хроматографического метода по сравнению с методами сорбции в статических условиях.
С помощью хроматографии возможны: разделение сложных смесей органических и неорганических веществ на отдельные компоненты, очистка веществ от примесей, концентрирование веществ из сильно разбавленных растворов, качественный и количественный анализ исследуемых веществ.
2.Классификация хроматографических методов
Воснову классификации многочисленных хроматографических методов положены следующие признаки:
1.агрегатное состояние фаз;
2.механизм взаимодействия сорбент – сорбат;
3.способы проведения хроматографического анализа;
4.аппаратурное оформление (техника выполнения) процесса хроматографирования;
5.цель хроматографирования.
По агрегатному состоянию фаз хроматографию разделяют на газовую и жидкостную. Газовая хроматография включает газожидкостную и газотвердофазную, жидкостная – жидкостно-жидкостную и жидкостно-твердофазную. Первое слово в названии метода
характеризует агрегатное состояние подвижной фазы, второе – неподвижной.
По механизму взаимодействия сорбента и сорбата можно выделить несколько видов хроматографии: адсорбционная основана на различии в адсорбируемости веществ твердым сорбентом; распределительная основана на различной растворимости разделяемых веществ в неподвижной фазе (газожидкостная хроматография) или на различной растворимости веществ в подвижной и неподвижной фазах (жидкостная хроматография); ионообменная хроматография – на разной способности веществ к ионному обмену; эксклюзионная хроматография – на различии в размерах и формах
молекул разделяемых веществ; аффинная хроматография – на специфических взаимодействиях, характерных для некоторых биологических и биохимических процессов (например, антитело и антиген, гормон и рецептор и др.). Существует осадочная хроматография, основанная на образовании отличающихся по растворимости осадков разделяемых веществ с сорбентом, адсорбционно-комплексообразовательная, основанная на образовании координационных соединений разной устойчивости в фазе или на поверхности сорбента, и др. Следует помнить, что классификация по механизму взаимодействия весьма условна: ее используют в том случае, если известен доминирующий механизм; часто процесс разделения протекает сразу по нескольким
механизмам.
По технике выполнения выделяют колоночную хроматографию, когда разделение проводится в специальных колонках, и плоскостную хроматографию, когда разделение проводится на специальной бумаге (бумажная хроматография) или в тонком слое сорбента (тонкослойная хроматография). В колоночной хроматографии используют насадочные или капиллярные колонки. Насадочную колонку заполняют сорбентом (насадкой), а внутреннюю стенку капиллярной колонки покрывают пленкой жидкости или
пылью адсорбента. |
|
|
|
|
|
|
|
В |
зависимости |
от цели |
проведения хроматографического |
процесса |
|||
различают аналитическую хроматографию |
(качественный |
и |
количественный |
||||
анализ); препаративнуюхроматографию (для получения веществ в чистом виде, для концентрирования и выделения микропримесей); промышленную (производственную) хроматографию для автоматического управления процессом (при этом целевой продукт из колонки поступает в датчик). Хроматографию часто используют для исследовательских целей при изучении растворов, каталитических процессов, кинетики химических
процессов и т.п.
Классификация по способам проведения анализа подразделяет хроматографию на
три вида: 1) фронтальный, 2) проявительный, 3) вытеснительный .
Фронтальный метод наиболее прост по выполнению. Через хроматографическую колонку с сорбентом непрерывным потоком пропускают раствор или газовую смесь исследуемых веществ, сорбируемость которых увеличивается в ряду А < В < С. Соответственно этому компоненты располагаются в колонке. Однако они разделяются не полностью. В чистом виде может быть выделен лишь первый, наиболее слабо сорбирующийся компонент, который движется вдоль слоя сорбента впереди остальных. За зоной первого компонента следует в непосредственном контакте зона, содержащая первый и второй компоненты. Третья зона содержит смесь первого, второго и третьего компонентов. В некоторый момент времени сорбент насыщается, и наступает «проскок», т.е. из колонки начинают выходить компоненты в соответствии с их сорбируемостью. Если пропускать жидкость или газ, выходящие из колонки, через детектор концентраций и наносить показания его в течение всего опыта на график, то полученная выходная кривая будет иметь форму ступенчатой кривой (рис.1.1).
Фронтальный метод не нашел широкого применения в анализе, т.к. не дает полного разделения компонентов анализируемой смеси. Однако этот метод весьма эффективен для препаративного выделения чистого вещества из технического образца при условии, что это вещество удерживается в колонке слабее всех других компонентов объекта анализа.
Типичные примеры применения фронтального анализа: очистка и умягчение воды ионообменными материалами; очистка воздуха активированными углями от отравляющих веществ в противогазах и вентиляционных фильтрах химических предприятий; концентрирование ценных веществ из сточных промышленных вод металлургических предприятий; очистка лекарственных препаратов и пищевых продуктов с помощью ионообменников и т.д.

Рис.. Выходная кривая фронтального анализа
А, В, С – разделяемые вещества
Проявительный (элюентный) метод выгодно отличается от фронтального тем, что он позволяет полностью разделить много-компонентную смесь. Хроматографическую колонку промывают растворителем или газом-носителем (элюентом), обладающим меньшей сорбируемостью, чем любое из разделяемых веществ. Затем в колонку вводят исследуемую смесь в виде порции раствора или газа, а не непрерывно, и продолжают пропускать элюент. При этом разделяемые вещества перемещаются вдоль колонки с разными скоростями в соответствии с их сорбируемостью. На выходе из колонки детектор фиксирует непрерывно концентрацию компонентов, а связанный с ним регистрирующий прибор записывает выходную кривую в виде ряда пиков, число которых соответствует числу разделенных компонентов Проявительный метод анализа получил широкое применение как в жидкостной, так и в
газовой хроматографии. Это объясняется тем, что при правильном выборе условий разделения компоненты смеси выходят из колонки в чистом виде, и их можно выделить для исследования другими методами анализа. Кроме того, качественный и количественный состав анализируемой смеси можно определить простым измерением объемов удерживания и площадей пиков соответствующих компонентов на полученной
хроматограмме.
Вытеснительный метод отличается от фронтального и проявительного тем, что после введения пробы исследуемой смеси колонку
Рис. 1.2. Выходная кривая проявительного анализа А, В, С – разделяемые вещества, Е – растворитель (элюент)
промывают растворителем или газом-носителем, к которым добавляют раствор вещества (вытеснитель), обладающего большей сорбируемостью, чем любое из разделяемых веществ. По мере продвижения по колонке элюент вытесняет вещество С, которое в свою очередь вытесняет вещество В и т.д. В результате вытесняемая смесь перемещается впереди фронта вытеснителя и скорость движения вещества равна скорости движения вытеснителя. Разделяемые вещества и на колонке, и в элюате располагаются последовательно друг за другом. Каждый из компонентов выделяется в чистом виде, но не количественно, так как зоны компонентов не разделены промежутками чистого сорбента. Невозможность получения на выходе из колонки достаточно чистых компонентов разделяемой смеси, а также длительность процесса разделения затрудняют использование
этого метода в аналитических целях. Однако для препаративных целей метод не потерял значения, так как возможность применения таких высокоактивных и доступных адсорбентов, как активированные угли, позволяет достигнуть высокой производительности. Достоинством метода является также то, что зоны не размываются в отличие от проявительного анализа.
1. 8 Лекция №8( 2 часа). Тема Жидкостная хроматография
1.8.1Вопросы лекции:
1.Жидкостная хроматография
1.8.2 Краткое содержание вопросов
Жидкостная хроматография-это метод разделения и анализа сложных смесей веществ, в котором подвижной фазой служит жидкость. Он применим для разделения более широкого круга веществ, чем метод газовой хроматографии. Это связано с тем, что большинство веществ не обладает летучестью, многие из них неустойчивы при высоких температурах (особенно высокомолекулярные соединения) и разлагаются при переводе в газообразное состояние. Разделение веществ жидкостной хроматографией чаще всего производится при комнатной температуре.− Особенности всех видов жидкостной хроматографии обусловлены тем, что подвижной
фазой в ней является жидкость, а сорбция компонентов из газообразного и жидкого элюента протекает по-разному. Если в газовой хроматографии газ-носитель выполняет только транспортную функцию и неподвижной фазой не сорбируется, то жидкая подвижная фаза в жидкостной хроматографии является активным элюентом, его молекулы могут сорбироваться неподвижной фазой. При прохождении через колонку молекулы компонентов анализируемой смеси, находящиеся в элюенте, должны вытеснить молекулы элюента из поверхностного слоя сорбента, что приводит к уменьшению энергии взаимодействия молекул анализируемого вещества с поверхностью сорбента. Поэтому величины удерживаемых объемов VR, пропорциональные изменению свободной энергии системы, в жидкостной хроматографии меньше, чем в газовой, а диапазон линейности изотермы сорбции в жидкостной хроматографии шире. природой подвижной (элюент) и неподвижной фаз.−Применяя различные элюенты, можно изменять параметры удерживания и селективность хроматографической системы. Селективность в жидкостной хроматографии в отличие от газовой определяется не одним, а двумя факторами Жидкая подвижная фаза имеет большую плотность и вязкость, чем газообразная,
коэффициенты диффузии Dж4 порядка ниже, чем в газе. Это приводит к замедлению массообмена в жидкостной хроматографии по сравнению с газовой. Уравнение ВанДеемтера в связи с тем, что член−на 3Вв жидкостной хроматографии роли не играет (Dж<<Dг), видоизменяется и графическая зависимость эффективностиНот линейной скорости потока подвижной фазы имеет вид, представленный на рис. 1.9.
100 мкм и элюентом, вводят анализируемую пробу, растворенную в элюенте, и пропускают элюент, отбирая на выходе из колонки порции элюата. Этот вариант жидкостной хроматографии до настоящего времени применяют в лабораторной практике, но так как скорость прохождения элюента под действием силы тяжести мала, анализ продолжителен.>В классическом варианте колоночной жидкостной хроматографии в стеклянную колонку высотой 1–2 м, заполненную сорбентом с размером частиц использует объемно- и поверхностно-пористые сорбенты с размером частиц 5–10 мкм, нагнетательные насосы, обеспечивающие давление в системе до 400 атм., высокочувствительные детекторы. Быстрый массоперенос и высокая эффективность разделения позволяют использовать ВЭЖХ для разделения молекул (жидкостноадсорбционная и жидкость-жидкостная распределительная хроматографии), для разделения ионов (ионообменная, ионная, ион-парная хроматография), для разделения макромолекул (эксклюзионная хроматографии).−так называемая высокоэффективная жидкостная хроматография ВЭЖХ−Современный вариант жидкостной хроматографии
УДЕРЖИВАНИЕ И СИЛА РАСТВОРИТЕЛЯ Для того чтобы анализируемые вещества разделялись на колонке, как уже упоминалось
выше, коэффициент емкости k' должен быть больше 0, т.е. вещества должны удерживаться неподвижной фазой, сорбентом. Однако коэффициент емкости недолжен быть и слишком большим, чтобы получить приемлемое время элюирования. Если для данной смеси веществ выбрана неподвижная фаза, которая их удерживает, то дальнейшая работа по разработке методики анализа заключается в выборе такого растворителя, который обеспечил бы в идеальном случае различные для всех компонентов, но приемлемо не очень большие k'. Этого добиваются, меняя элюирующую силу растворителя.
В случае адсорбционной хроматографии на силикагеле или оксиде алюминия, как правило, силу двухкомпонентного растворителя (например, гексана с добавкой изопропанола) увеличивают, увеличивая в нем содержание полярного компонента (изопропанола), или же уменьшают, уменьшая содержание изопропанола. Если содержание полярного компонента становится слишком малым (менее 0,1%), следует заменить его более слабым по элюирующей силе. Так же поступают, заменяя на другие либо полярную, либо неполярную составляющую и в том случае, если данная система не обеспечивает желаемой селективности по отношению к интересующим компонентам смеси. При подборе систем растворителей принимают во внимание как растворимости
компонентов смеси, так и элюотропные ряды растворителей, составленные разными авторами.
Примерно так же подбирают силу растворителя в случае использования привитых полярных фаз (нитрил, амино, диол, нитро и др.), учитывая возможные химические реакции и исключая опасные для фазы растворители (например, альдегиды и кетоны для аминофазы).
В случае обращенно-фазной хроматографии силу растворителя увеличивают, повышая содержание в элюенте органической составляющей (метанола, ацетонитрила или ТГФ) и уменьшают, добавляя больше воды. Если не удается добиться желаемой селективности, используют другую органическую составляющую либо пытаются изменить ее с помощью разных добавок (кислот, ион-парных реагентов и др.).
При разделениях методом ионообменной хроматографии силу растворителя меняют, увеличивая или уменьшая концентрацию буферного раствора или меняя рН, в некоторых случаях используют модификацию органическими веществами.
Однако, особенно в случае сложных природных и биологических смесей, зачастую не удается подобрать силу растворителя таким образом, чтобы все компоненты пробы элюировались за приемлемый срок. Тогда приходится прибегать к градиентному элюированию, т.е. использовать растворитель, элюирующая сила которого в процессе анализа изменяется так, что она постоянно увеличивается по заранее заданной программе. Таким приемом удается добиться элюирования всех компонентов сложных смесей за относительно короткий промежуток времени и их разделения на компоненты в виде узких
пиков.
Адсорбционная жидкостная хроматографиянеполярный адсорбент и полярные подвижные фазы, но в обоих вариантах выбор подвижной фазы часто важнее, чем выбор неподвижной. Неподвижная фаза должна удерживать разделяемые вещества. Подвижная фаза, т. е. растворитель, должна обеспечивать различную емкость колонки и эффективное разделение за приемлемое время.−. Адсорбционная жидкостная хроматография в зависимости от полярности неподвижной и подвижной фаз подразделяется на нормальнофазовую (НФХ) и обращенно-фазовую (ОФХ) хроматографии. В НФХ используют полярный адсорбент и неполярные подвижные фазы, в ОФХ
В качестве неподвижной фазы в адсорбционной жидкостной хроматографии применяют полярные и неполярные тонкодисперсные пористые материалы с удельной поверхностью более 50 м2/г. Полярные адсорбенты (SiO2,Al2O3, флорисил и др.) имеют на поверхности слабокислотные гвысокая чувствительность к содержанию воды в применяемых элюентах. Для устранения этого недостатка полярные сорбенты