

Способы выражения концентрации (процентная, молярная, эквивалентная). Связь между концентрациями.
Для количественной характеристики растворов используют понятие концентрации:
Концентрация – величина, выражающая относительное содержание данного компонента в системе (смеси, растворе).
Из концентраций растворов наибольшее применение в химии находят следующие:
Процентная концентрация растворов показывает число единиц массы растворенного вещества, содержащееся в 100 единицах массы раствора, и для его приготовления следует взять 12 единиц массы СаСl2 и 88 единиц массы растворителя.
Молярная концентрация раствора (молярность) – отношение количества этого вещества, содержащегося в растворе (в молях), к объему раствора:
![]() ,
,
где m – масса растворенного вещества, г; М – молярная масса растворенного вещества, г∙моль-1; V – объем раствора, л. Единица Си – моль∙м-3, обычно применяют моль∙л-1.
Молярным называется раствор, в 1л которого содержится 1 моль растворенного вещества.
Эквивалентная (нормальная) концентрация раствора (нормальность) – отношение числа эквивалентов вещества, содержащегося в растворе, к объему раствора:
![]()
где m – масса растворенного вещества; Мэкв – молярная масса эквивалента растворенного вещества.
Единица эквивалентной концентрации в СИ – моль∙м-3, обычно применяют моль∙л-1.
Молярная концентрация раствора (моляльность) определяется числом молей растворенного вещества в 1кг (1000г) растворителя. Единица моляльности раствора в СИ – моль∙кг-1, можно применять моль∙г-1.
Основная особенность моляльного способа выражения концентрации заключается в том, что моляльная концентрация раствора не зависит от температуры, поскольку для определения моляльности не привлекается объем.
Массовой долей растворенного вещества называют отношение массы растворенного вещества к общей массе раствора. Массовую долю обычно выражают в долях единицы и обозначают W.
Мольная доля – отношение числа молей данного вещества в растворе к общему числу молей веществ, образующих раствор.
Для приготовления растворов заданных концентраций должны проводиться соответствующие расчеты.
Зная плотность раствора, можно легко установить взаимосвязь между различными способами выражения его концентрации. Для этого необходимо решить в общем виде систему уравнений, состоящую из формул, выражающих его концентрации.
Понятие «эквивалент». Закон эквивалентов. Применение закона эквивалентов в объемном анализе.
Одним из основных законов химии является закон эквивалентов, открытый в конце 18 века: массы элементов, соединяющихся друг с другом, пропорциональны их эквивалентам:
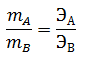
где mA, mВ – массы взаимодействующих веществ А и В;
ЭА и ЭВ – эквиваленты этих веществ.
Эквивалент – это реальная или условная частица, которая в кислотно-основных реакциях ионного обмена равноценна одному атому или одному иону водорода, а в окислительно-восстановительных реакциях одному электрону.
Массу одного моля эквивалента элемента называют молярной массой эквивалента MЭ(X). Значение эквивалента веществ зависит от того, в какой конкретной реакции участвует это вещество.
Молярная масса эквивалента химического элемента (MЭ(X)), а также простых или сложных веществ рассчитывается по формуле
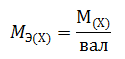
где M(X) – молярная масса; вал – суммарная валентность.
Для простых веществ суммарная валентность определяется произведением валентности атома химического элемента и числа атомов. Так, молярная масса эквивалента алюминия составляет MЭ(AI) = 27/3 = 9г моль-1. Молярные массы эквивалента кислорода и водорода следует запомнить, они равны соответственно MЭ(О) = 16/2=8г моль-1, МЭ(Н) = 1/1 = 1 г моль-1.
Молярные массы эквивалента сложных веществ вычисляются по их химическим формулам с учетом происходящих химических реакций.
К сложным веществам относятся оксиды, гидроксиды, соли.
Суммарная валентность оксидов равна произведению валентности кислорода (2) на количество атомов кислорода в молекуле. Суммарная валентность гидроксидов определяется их кислотностью, которая равняется числу замещенных гидроксильных групп. Суммарная валентность кислот равняется основности данных соединений, которая определятся числом замещенных атомов водорода. Суммарная валентность соли равняется произведению валентности катиона и количества катионов в молекуле, или валентности аниона и количества анионов в молекуле.
Молярная масса эквивалентов сложных веществ может быть определена как сумма молярных масс эквивалентов элементов или условных частиц, образующих данное вещество.
Мэ (оксиды) = Мэ (О) + Мэ (элемента) = 8 + Мэ (элемента), т.к. Мэ кислорода величина постоянная, равна 8г моль-1.
Мэ (кислота) = Мэ (Н) + Мэ (кислотного остатка) = 1 + Мэ (кислотного остатка), т.к. Мэ водорода величина постоянная, 1г моль-1
Мэ (гидроксиды) = Мэ (OH-) + Мэ (металла) = 17 + Мэ (металла), т.к. Мэ “OH” групп величина постоянная, равна 17г моль-1.
Мэ (соли) = Мэ (катиона) + Мэ (кислотного остатка).
Фактор эквивалентности fэ (х) – число, обозначающее, какая доля реальной частицы вещества X эквивалентна одному иону водорода в кислотно-основной реакции или одному электрону в реакции окисления-восстановления.
Фактор эквивалентности – величина безразмерная. Принимает значения 1 или меньше единицы.
Для простых веществ и элементов в соединении fэ(х) = 1/В, где В – валентность элемента.
Например, для водорода или натрия fэ= 1/1 = 1. Для магния или кислорода fэ = 1/2.
Объемный анализ основан на законе эквивалентов: эквивалентные количества всех веществ, участвующих в реакции, одинаковы.
Для реакции aA + bB = cC + dD в соответствии с законом эквивалентов всегда будет справедливо равенство:
neqA = neqB = neqC = neqD
Поэтому, если эквивалентное количество одного из веществ (реагента или продукта) известно, то определены и эквивалентные количества всех остальных веществ, участвующих в данной реакции, а необходимость их расчета отпадает. В этом состоит преимущество проведения стехиометрических расчетов по закону эквивалентов.
Качественный и количественный анализ в аналитической химии.
Аналитическую химию разделяют на две основные части: качественный и количественный анализ. Деление на качественный и количественный анализ в какой-то степени условно и традиционно. Задача качественного анализа – обнаружить, какие именно элементы или их соединения входят в состав анализируемого материала.
Качественный анализ обычно предшествует количественному; цель последнего – найти количественные соотношения между компонентами, найденными при качественном исследовании.
Идентификация компонентов и определение качественного состава объекта являются предметом качественного анализа. Качественный анализ, как мы уже указывали, обычно предшествует количественному.
При обнаружении какого-либо компонента обычно фиксируют появление аналитического сигнала: образование осадка, изменение окраски, выделение газа, появление линий в спектре и т. д.
Для получения сигнала в аналитической химии используют химические реакции разных типов (кислотно-основные, окислительно- восстановительные, комплексообразования), разные процессы, на- пример осаждение, а также разнообразные химические, физические и даже биологические свойства самих веществ или продуктов их реакций. Поэтому аналитическая химия располагает различными методами для решения своих задач: химическими, физическими и биологическими.
В качественном химическом анализе преимущественно имеют дело с водными растворами электролитов, поэтому аналитическими реакциями открывают образующиеся при диссоциации ионы.
Для удобства обнаружения ионы делятся на аналитические группы.
При объединении ионов в группы используют сходство или различие их свойств в отношении действия некоторых реактивов, называемых групповыми, различную растворимость образуемых ими соединений и другие признаки.
Методами количественного химического анализа устанавливают концентрации или количества составных веществ, а также в каких количественных соотношениях они находятся в исследуемом объекте.
Количественному анализу обычно предшествует выяснение качественного состава образца, так как важно не только подтвердить наличие в образце того или иного компонента, но и выяснить, какие другие вещества сопутствуют определяемому соединению.
Методы количественного определения принято разделять на химические, физико-химические, физические и биологические. Приведенная классификация достаточно условна, поскольку четкого разграничения между названными методами провести нельзя.
Химические или, как их еще называют, классические методы анализа базируются на химических реакциях. В химических методах количественного анализа проводят химическую реакцию и измеряют либо массу полученного продукта (гравиметрия), либо затраченный объем реагента (титриметрия), что дает возможность судить о количестве определяемого вещества.
Общая характеристика инструментальных методов анализа. Классификация инструментальных методов анализа.
Классические методы химического анализа : гравиметрический метод(масса осадка - аналит сигнал), титриметрический анализ(объем титранта –аналит сигнал). Однако химические методы анализа не в состоянии удовлетворить многообразные запросы практики
Инструментальный м-д –характеристики:
очень быстро снижают предел обнаружения(до 10-5-10-10 %),
экспрессность (3 параллельной пробы, высокий темп получения результата),
возможность дистанционного анализа(пр-р анализ лунного грунта),
автоматизация процесса анализа(пр-р газоанализаторы, контролируют состав воздуха в шахтах),
анализ с помощью некоторых физ –хим методов может быть выполнен без разрушения анализируемого образца.
Достоинства :
Низкий предел обнаружения(1-10-9мкг)
Малая предельная концентрация(10-12-10-15гр/мл определяемого в-ва)
Высокая чувствительность (формально опред-мая тангенсом угла наклона- градуировочноая
Г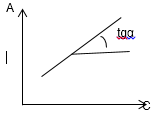
 радуировочная
кривая отображает график зависимости
физ-го параметра(аналит. сигнала) от
концентрации или количества определяемого
в-ва. Концентрация по оси Х ,по оси
У-сигнал .чем больше тангенс угла наклона
,тем чувствителен метод.
радуировочная
кривая отображает график зависимости
физ-го параметра(аналит. сигнала) от
концентрации или количества определяемого
в-ва. Концентрация по оси Х ,по оси
У-сигнал .чем больше тангенс угла наклона
,тем чувствителен метод.
Высокая селективность(избирательность)(она позволяет определить соответствующий компонент без предварительного разделения смеси на компоненты
Недостатки:
Иногда воспроизводимость может быть хуже химического анализа
Погрешность измерения (чаще 5-10%,иногда 20%)
Сложность аппаратуры и ее высокая стоимость
Необходимость исп-я эталонов,стан-х р-в,градуировочного графика
Классификакция:
Оптические м-ды основаны на из-и оптических св-в
Хроматографические методы основан на использовании способности разных в-в к избирательной сорбции
Электро-хим. методы основаны на электрических св-х системы
Радиометрические основаны на изменении радиоактивных св-в в-в
Термические методы основаны на изменении тепловых эффектов
Масс-спектометрический метод основан на способности газооб-х ионов разделяться в магнитном поле в зависимости от отношения массы к заряду иона.
Классификация и характеристика спектральных методов анализа.
Оптические методы анализа основаны на измерении оптических свойств веществ, которые проявляются при взаимодействии эл/м излучения с веществом.
Оптические свойства: испускание, поглощение, рассеяние, отражение, преломление, поляризация света.
Классификация:
а) по изучаемым объектам:
1. атомный спектральный анализ;
2. молекулярный спектральный анализ.
б) по характеру взаимодействия эл/м излучения с веществом:
1. Атомно-адсорбционный метод: в основе метода лежит измерение поглощения монохроматического излучения атомами определяемого вещества в газовой фазе после его атомизации;
2. Эмиссионный спектральный анализ: атомный метод анализа, основанный на изменении интенсивности света, излучаемого веществом при его энергетическом возбуждении.
3. Пламенная фотометрия. Основана на использовании газового пламени в качестве источника энергетического возбуждения излучения.
4. Молекулярная абсорбционная спектроскопия. Основана на измерении светопоглощения молекулами или ионами изучаемого вещества.
5. Люминесцентный анализ – определение/измерение интенсивности люминесценции.
6. Спектральный анализ с эффектом комбинационного рассеяния света (рамановская спектроскопия). Рассматривает рассеяние света.
7. Нефелометрический анализ. Основан на измерении рассеивания света частицами дисперсной среды.
8. Турбидиметрический анализ. Измерение ослабления интенсивности излучения при прохождении через дисперсную среду.
9. Рефрактометрический анализ. Измерение показателя преломления света.
10. Интерферометрический метод анализа.
11. Поляриметрический метод анализа. Измерение величины оптического вращения, а именно угла вращения плоскости поляризации света оптически активными веществами.
в) по области использования эл/м спектра
1. -спектроскопия в ближней УФ области (200-400нм);
- спектроскопия в видимой области оптического диапазона (400-750нм);
2. ИК спектроскопия (750-105нм);
3. Рентгеновская спектроскопия.
г) по природе энергетических переходов:
Электронный спектр;
Колебательный спектр;
Вращательный спектр.
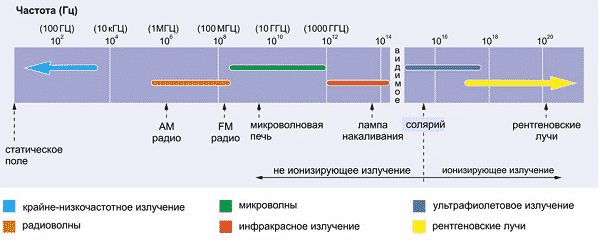
Основные характеристики электромагнитного излучения: Шкала электромагнитного излучения.
Поскольку свет имеет двойственную природу, то для его описания используют 2 вида характеристик.
Волновые: частота, длина волны, волновое число.
Квантовые: энергия квантов.
Частота показывает число колебаний в 1 с.
Длина волны показывает наименьшее расстояние между точками, колеблющимися в одной фазе.
Окраска вещества связана с избирательным светопоглощением. Если вещество поглощает свет, то оно может быть окрашено. Свет, который не поглотился, воспринимается нами, и мы видим какой-то цвет.
Цвет – свойство света вызывать определенные зрительные ощущения в соответствии со спектральным составом отражаемого или испускаемого излучения.
Длина волны и частота связаны следующим соотношением: 𝜈=𝑐𝜆
Волновое число используется при характеристике ИК-спектра: 𝜈̃=1𝜆
Энергия кванта света: 𝐸=ℎ𝜈
*Таблица дополнительных цветов.
Длины электромагнитных волн, которые могут быть зарегистрированы приборами, лежат в очень широком диапазоне. Все эти волны обладают общими свойствами: поглощение, отражение, интерференция, дифракция, дисперсия. Свойства эти могут, однако, проявляться по-разному. Различными являются источники и приемники волн.
Радиоволны
ν=105- 1011 Гц, λ=10-3-103 м.
Получают с помощью колебательных контуров и макроскопических вибраторов. Свойства. Радиоволны различных частот и с различными длинами волн по-разному поглощаются и отражаются средами. Применение Радиосвязь, телевидение, радиолокация. В природе радиоволны излучаются различными внеземными источниками (ядра галактик, квазары).
Инфракрасное излучение (тепловое)
ν=3-1011- 4.1014 Гц, λ=8.10-7 - 2.10-3 м.
Излучается атомами и молекулами вещества.
Инфракрасное излучение дают все тела при любой температуре.
Человек излучает электромагнитные волны λ≈9.10-6 м.
Свойства
Проходит через некоторые непрозрачные тела, а также сквозь дождь, дымку, снег.
Производит химическое действие на фотопластинки.
Поглощаясь веществом, нагревает его.
Вызывает внутренний фотоэффект у германия.
Невидимо.
Регистрируют тепловыми методами, фотоэлектрическими и фотографическими.
Применение. Получают изображения предметов в темноте, приборах ночного видения (ночные бинокли), тумане. Используют в криминалистике, в физиотерапии, в промышленности для сушки окрашенных изделий, стен зданий, древесины, фруктов.
Видимое излучение
Часть электромагнитного излучения, воспринимаемая глазом (от красного до фиолетового):
![]()
Свойства. Воздействует на глаз.
Ультрафиолетовое излучение
![]()
(меньше, чем у фиолетового света)
Источники: газоразрядные лампы с трубками из кварца (кварцевые лампы).
Излучается всеми твердыми телами, у которых T>1000°С, а также светящимися парами ртути.
Свойства. Высокая химическая активность (разложение хлорида серебра, свечение кристаллов сульфида цинка), невидимо, большая проникающая способность, убивает микроорганизмы, в небольших дозах благотворно влияет на организм человека (загар), но в больших дозах оказывает отрицательное биологическое воздействие: изменения в развитии клеток и обмене веществ, действие на глаза.
Рентгеновские лучи
Излучаются при большом ускорении электронов, например их торможение в металлах. Получают при помощи рентгеновской трубки: электроны в вакуумной трубке (р= 10-3-10-5 Па) ускоряются электрическим полем при высоком напряжении, достигая анода, при соударении резко тормозятся. При торможении электроны движутся с ускорением и излучают электромагнитные волны с малой длиной (от 100 до 0,01 им). Свойства Интерференция, дифракция рентгеновских лучей на кристаллической решетке, большая проникающая способность. Облучение в больших дозах вызывает лучевую болезнь. Применение. В медицине (диагностика заболеваний внутренних органов), в промышленности (контроль внутренней структуры различных изделий, сварных швов).
γ-излучение
Источники: атомное ядро (ядерные реакции). Свойства. Имеет огромную проникающую способность, оказывает сильное биологическое воздействие. Применение. В медицине, производстве (γ-дефектоскопия). Применение. В медицине, в промышленности.
Общим свойством электромагнитных волн является также то, что все излучения обладают одновременно квантовыми и волновыми свойствами. Квантовые и волновые свойства в этом случае не исключают, а дополняют друг друга. Волновые свойства ярче проявляются при малых частотах и менее ярко - при больших. И наоборот, квантовые свойства ярче проявляются при больших частотах и менее ярко — при малых. Чем меньше длина волны, тем ярче проявляются квантовые свойства, а чем больше длина волны, тем ярче проявляются волновые свойства.
Основные узлы спектральных приборов, используемых в спектрофотометричеком методе анализа.
Прибор для проведения спектрального ан-за имеет следующие основные узлы: источник возбуждения, диспергирующий элемент и приемник света. Всегда есть оптическая система, предназначенная для получения парал-го пучка света, его фокусировки, изменения хода лучей и т. д. В ист-ке возбуждения в-во атомизируется и возбужденные атомы или ионы испускают свет, который диспергирующим элементом разделяется в пространстве на отдельные составляющие, а приемник света их фиксирует.
Источники возбуждения переводят пробу из конденсир-ой фазы в парообразную и возбуждают в-во в этой фазе. В большинстве источников возбуждения эти функции совмещаются, но в иногда применяют два устройства: одно для получения газовой фазы, другое — для возбуждения. Возбуждение атомов происходит при передаче энергии быстролетящими частицами, чаще всего электронами, если их энергия достаточна для возбуждения. Если кинетическая энергия летящих частиц меньше энергии возбуждения первого возб-го уровня, при столкновении произойдет лишь перераспр-ие энергии, как при ударе упругих шаров, но возбуждения не произойдет(- это упругие соударения.) Чтобы атом перешел в возбужденное состояние, нужна энергия, равная хотя бы энергии резонансного уровня атома. Соударения, сопровожд-ся возбуждением атома, называются неупругими ударами первого рода.
К источнику возбуждения относят и устройство для введения анализируемой пробы. Р-ры вводят в источник возбуждения с помощью распылителей, порошкообразные пробы — с помощью специальных устройств или при использовании угольных эл-дов, в кот-х высверливается канал для набивки порошкообразной пробы
Пламя.. дает достаточно яркий и стабильный спектр. Простота регулировки и надежность работы пламенных источников. Возбуждение спектров в пламени имеет термический характер. Может быть газовая горелка работающая на смеси водорода и воздуха, кислорода и водорода, ацетилена и кислорода. С помощью пламенных источников определяют свыше 40 элементов (Mg, Сu, Mn, Т1, щелочные элементы, щелочно-земельные и т. д.).
Дуга. Электрическая дуга — это электрический разряд при большой силе тока (5...7 А) и небольшом напряжении (50...80 В). Разряд поддерживается за счет термоэлектронной эмиссии с раскаленной поверхности катода. Разряд пропускают между электродами из анализ-го образца или между образцом и эл-дом, не содержащим опр-х элементов. Для ан-за легкоплавких Ме, сплавов и руд, минералов и др. непроводящих мат-лов электродами служат графитовые или угольные стержни — так называемые спектральные угли.
Искра. Для ее получения используют искровые генераторы. Яркость искрового спектра недостаточна для визуального анализа. Достоинство : большая стабильность условий разряда и необходимая в колич-ном анализе стабильность условий возбуждения. Работа с искрой практически не вызывает разрушения образца, в отличие от дуги. Перспективным высокочувст-ым источником света явл-ся также полый катод, в котором могут возбуждаться элементы с высоким потенциалом возбуждения.
Диспергирующий
элемент .
Разлагает излучение в спектр. Он хар-ся
угловой дисперсией, которую определяют
как угловое расстояние ∆ϕ между двумя
лучами с близкими длинами волн λ1
и λ2,
отнесенное к интервалу ∆λ=
λ1 -
λ2,
т.е. Dϕ=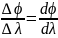 .
От угловой дисперсии
диспергирующего элемента зависит
линейная дисперсия спектрального
прибора Dl=
.
От угловой дисперсии
диспергирующего элемента зависит
линейная дисперсия спектрального
прибора Dl=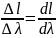 .
где ∆I
—линейное расстояние в фокальной
плоскости прибора между двумя лучами
с близкими длинами волн λ1
и λ2,
отнесенное к разности
∆λ=λ1
- λ2
. Разрешающей
способностью прибора- сп-ть давать
раздельное изображение двух спектр-х
линий с близкими длинами волн.
В качестве дисперг-го
элемента исп-т призмы, дифракц-ые решетки
и интерференционные устройства.
.
где ∆I
—линейное расстояние в фокальной
плоскости прибора между двумя лучами
с близкими длинами волн λ1
и λ2,
отнесенное к разности
∆λ=λ1
- λ2
. Разрешающей
способностью прибора- сп-ть давать
раздельное изображение двух спектр-х
линий с близкими длинами волн.
В качестве дисперг-го
элемента исп-т призмы, дифракц-ые решетки
и интерференционные устройства.
Приемники света. Хар-тся спектральной чувств-тью: сп-тью воспринимать излучение различной длины волны и интегральной чувств-тью, которая измеряется действием неразложенного в спектр излучения. Более универсальными приемниками света являются фотопластинки и фотоэлементы.
Фотопластинка. Светочувствительный слой фотопластинки — это мелкие кристаллы галогенидов серебра, равномерно распределенные в тонком желатиновом слое. Другим важным свойством фотопластинки является ее чувствительность.
Фотоэлементы. устройства, преобразующие световую энергию в электрическую. Его действие основано на использовании фотоэффекта. Различают внешний и внутренний фотоэффекты. При внешнем фотоэффекте поглощение света приводит к отрыву электрона с облучаемой поверхности. Внутренний фотоэффект хар-ся увеличением электрической проводимости в-ва под действием света. Если внутренний фотоэффект проявляется вблизи граничного слоя между двумя полупроводниками или полупроводником и металлом, то возникает фотоЭДС. Это явление иногда выделяют в особый вид фотоэффекта и называют фотогальваническим эффектом или эффектом запорного (запирающего) слоя.
Абсорбционная спектроскопия. Основной закон светопоглощения. Спектры поглощения.
Абсорбционная спектроскопия основана на изучение спектра поглощения анализ-го в-ва .Каждое в-во поглощает или отражает лучи света. Величина поглощения света опр-тся природой ана-го в-ва и его конц-ии в р-ре. Возможен кач-ый и колич-ый ан-з. Абсорбционная анализ подразделяется на атомный и молекулярный.
Основной закон светопоглощения:
Закон Бугера-Ламберта: Каждый тонкий слой постоянной толщины внутри однородной среды поглощает одинаковую долю падающего на него светового потока. Доля светового потока поглощенного однородной средой прямо пропорц-но толщине поглощательного света.
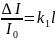 ; где l
–толщина поглощающего света. ∆I=I0*I;
где I-инст-ть
илз-я пад-го на среду , I0
–инт-ть пад-го света
; где l
–толщина поглощающего света. ∆I=I0*I;
где I-инст-ть
илз-я пад-го на среду , I0
–инт-ть пад-го света
 Закон
Бугера-Бера:
для светового потока ,поглощающего
данным тонким слоем внутри однородной
среды, прямо пропорциональна концентрации
поглощающих частиц.
Закон
Бугера-Бера:
для светового потока ,поглощающего
данным тонким слоем внутри однородной
среды, прямо пропорциональна концентрации
поглощающих частиц.
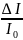 =k2C
=k2C
Объединенный закон: связывает уменьшение интенсивности света поглощенного через слой светопоглощенного вещества с концентрацией этого вещества и толщиной слоя.
Физический смысл оптической плотности –отрицательный логарифм. Физический смысл молярного коэф-та поглощения-равен оптической плотности одномерного раствора с толщиной поглощаемого света в 1 см.
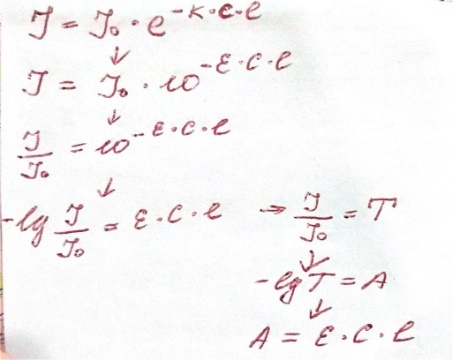

Свет поглощается раствором избирательно: при некоторых длинах волн светопоглощение происходит интенсивно, а при некоторых свет не поглощается. Интенсивно поглощаются кванты света, энергия которых hx равна энергии возбуждения частицы и вероятность их поглощения больше нуля. Распределение по частотам (или по длинам волн) значений молярного коэффициента поглощения называется спектром поглощения. Его выражают в виде графической зависимости оптической плотности А или молярного коэф-та поглощения е от частоты v или длины волны к падающего света. Вместо А или е нередко откладывают их логарифмы.
Основные узлы: источник света, монохроматизатор света, кювета с исслед-м в-вом, рецептор (приемник света).Так же оптическая система, состоящая из линз, призм и зеркал, которая служит для создания парал-го пучка света, изменения направления и фокусировки света, а также систему для уравнивания интенсивности световых потоков (диафрагмы и т. д.).
Основными источниками освещения являются вольфрамовые лампы накаливания, газонаполненные лампы (водородная, ртутная), штифт Нернста и глобар. В лампе накаливания светящаяся вольфрамовая спираль дает свет в широком спектральном интервале. В водородной лампе происходит свечение водорода при разряде. В ртутной лампе разряд происходит в парах ртути. Штифт Нернста - столбик, спрессованный из оксидов редкоземельных элементов. Глобар-штифт из карборунда SiC дает излучение.
Монохроматизаторами или монохроматорами называют устройства для получения света с заданной длиной волны. При конструировании монохроматизаторов используют разные оптические явления: поглощение света, интерференцию, дисперсию и т. д. В практике имеют приборы, в которых в качестве монохроматизаторов применяются светофильтры и призмы, дифрак-е решетки.
Типы светофильтров: абсорбционные, интерференционные или интерференционно-поляризационные. Действие абсорбц-х основано на прохождении света через тонкий слой вследствие поглощения происходит изменение величины и спектрального состава проходящего светового потока. Интерференционный состоит из двух тонких полупрозрачных слоев серебра, между кот-ми находится слой диэлектрика. Универсал-ми монохроматизаторами являются призмы, изготовл-ые из кварца, стекла и некоторых других материалов.
Кюветное отделение рассчитано на какое-то количеств кювета.
В кач-ве рецепторов в приборах абсорбц-ой спектр-пии исп-т главным образом фотоэл-нты, фотоумн-ли, а иногда инт-ть света оценивается на глаз. Для измерения инт-ти инфракр-го излучения прим-т фотоэл-нты, термоэл-нты и болометры. Приемники света хар-ся спектральной чувств-тью — сп-тью воспринимать излучение различной длины волны — и интегральной чувств-тью, которая измеряется по действию на рецептор не разложенного в спектр излучения. В термоэл-нтах исп-тся термоЭДС, возникающая при изменении темп-ры спая между металлами или сплавами под действием инфракр-го излучения.
Ограничения и условия применения закона Бугера-Ламберта-Бера. Положительные и отрицательные отклонения от закона Б-Л-Б. Физико-химические и инструментальные причины отклонений.


Ограничение закона Б-Л-Б.
Свет должен быть строго монохроматичным
Коэффициент поглощения зависит от показателя преломления исследуемой стреды
Температура должна оставаться постоянной
Пучок света должен быть параллельным
Интенсивность рассеянного света должна быть сведена к минимуму.
Отклонение могут быть обусловлены: физико-химическими и инструментальными причины.
Физико-химические: при увеличении концентрации раствора после некоторых величин предельной концентрации становится заметны процессы ассоциации, И т.д. при этом число светопоглощающих частиц может уменьшаться, а следовательно оптическая плотность падает. Поглощательное отношения связно с тем ,что новые образовавшиеся частицы могут обладать большей светопоглощающей способностью ,чем исходный частицы при данной длине волны.
Иногда наблюдается более сложное отклонение от линейной зависимости оптической плотности концентрации. Кажущиеся отклонения могут наблюдаться при уменьшении концентрации раствора тоже за счет процессов диссоциации и так далее.
Инструментальные отклонения: связанное преимущественно с недостаточностью монохроматичностью светового потока, влияние света рассеивания ,нелинейность работы приемников излучения. Указанное отклонение называется кажущиеся поскольку закон света поглощения не нарушается, либо изменяется число светопоглощающих частицы, либо меняется природа светопоглощающих частиц, либо спектральный прибор точно регистрируют интенсивность светового потока, либо спектральный прибор не точно измеряют интенсивность светового потока прошедшего через раствор.
На практике в начале на основании изменения оптической плотности эталоных растворов и с изменением концентрации определяемого вещества устанавливается предел изменение концентрации при которых выполняется закон светопоглощения, а далее проводят изменения оптической плотности анализируемых растворов в данном концентрационном диапазоне и в тех же условиях. При наличии экспериментальной установленной зависимости оптической плотности от концентрации можно проводить аналитические измерения и без строгого соблюдения закона, а пользовавшись градуированным графиком. Не рекомендуется проводить измерения оптических плотности при очень малых и очень больших величинах ,так как при этом резко возрастает ошибка измерений.
Спектрофотометрический метод анализа. Основные приемы спектрофотометрических измерений.
Спектрофотометрия изучает поглощение анализируемым веществом света с определённой длиной волны(монохроматическое излучение) . Прибор-спектрофотометр. Узлы спректрофотометра –источник излучения, свет из источника излучения попадает в монохроматы. У веществ в зависимости от их природы наиболее яркие полосы- спектральные ,располагаются в разных частях спектра.
Любое спректрофотометрическое определние состоит в основном из двух этапов:
Проведение с испытуемым р-ром хим. реакций для получения систем пригодных для фотометрии.
Измерение поглощения приготовленных растворов.
Для проведения определяемого вещества в соединение поглощения используют реакции комплексообразования, реакции ионного обмена, ОВР. Условия проведения реакции должны быть изучены, т.к. они должны обеспечить воспроизводимость и наклонность спектрометрического определения.
Оптимальные условия фотометрического определения:
1.Длина волны. При определении в растворе одного светопоглощающего вещества аналитическую длину волны выбирают на максимуме полосы поглощения. Если в спектре имеется несколько полос, выбор обычно останавливают на наиболее интенсивной, так как работа в области максимума светопоглощения обеспечивает наиболее высокую чувствительность определения. Плоские максимумы более предпочтительны, так как при этом меньше сказывается погрешность в установлении длины волны, чем в случае острых максимумов или крутоспадающих участков кривой. Желательно также, чтобы чувствительность приемника излучения в области аналитической длины волны была максимальна.
2 .Диапозон
концентрации и толщины поглощательного
света; выбор толщины и концентрации так
же является важным условием проведения
анализа.
.Диапозон
концентрации и толщины поглощательного
света; выбор толщины и концентрации так
же является важным условием проведения
анализа.
Светопропускание (оптическая плотность). Измерительное устройство фотометрического прибора обычно имеет постоянную ошибку ∆Т в величине коэффициента пропускания Т во всем интервале его значений. Фотометрическое измерение целесообразно проводить от 0,2-0,8.Поэтому до начала проведения анализа готовят серию станд-х растворов с различной концентрацией определяемого вещества, находят предел изменения концентрации и оптических плотностей в которых выполняется закон светопоглощения.
3.Раствор сравнения(нулевой р-р)-это либо чистый р-ль,если измеренный раствор состоит из р-ля и раств-го в-ва, раст-ль содержащий все те же компоненты и в тех же кол-х , что и измеренный р-р ,за искл-ем определяемого вещества. Все последующие измерения проводят по отношению к р-ру сравнения. Измерения следует проводить достаточно быстро, так как при продолжительном нахождении в кюветном отделении кюветы с р-м нагреваются ,при этом возможно появление мелких пузырьков воздуха на стенках кювета, что искажает работу спектрофотометрического измерения.
4.Чувствительность метода: Минимальную концентрацию, которую можно определить фотометрическим методом, обычно рассчитывают по соотношению Сmin=Amin/(εl).ε является основной характеристикой чувствительности метода. Точность фотометрических методов зависит от индивидуальных особенностей фотометрической реакции, характеристик используемого прибора и тд. Точность изменяется в широком интервале (1-2%).
Основные приемы фотометрических измерений:
Метод градуировочного графика: готовят 5-6 эталонных р-в с различной концентрацией определяемого вещества по реакциям измерения оптической плотности эталонных растворов при выбранной длине аналитической волны стоится график А от С. Измеряют А(х) анализируемого раствора с неизменной концентрацией определяемого вещества. В тех же условиях по графику находят С(х) соответствующий А(х),дальше ведут пересчет с учетом всех этапов разбавления .Графический способ применяют тогда, когда наблюдается кажущееся отклонение от основного закона светопоглощения.
Метод одного стандарта: применяется тогда, когда точно выполняется закон светопоглощения. Готовим стандартный р-р с измеренной концентрацией определяемого вещества .
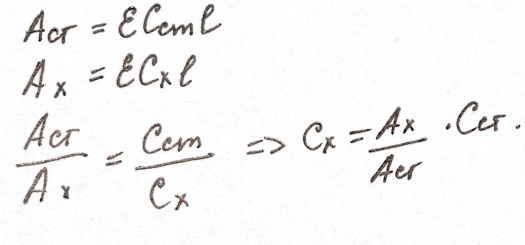 Метод
молярного коэффициента поглощения:
Определяет
оптическую плотность нескольких
стандартных растворов ε=Аст/Сстl
. Получение значения
ε усредняют, измеряют А анализируемого
раствора, делают обратную расчетную
операцию Сх=Ах/εl
. Ограничения:
обязательное подчинение закону
светопоглощения для аналитической
системы, т.е. концентрация анализируемого
раствора должна попасть во внутрь
диапазона концентрации раствора.
Метод
молярного коэффициента поглощения:
Определяет
оптическую плотность нескольких
стандартных растворов ε=Аст/Сстl
. Получение значения
ε усредняют, измеряют А анализируемого
раствора, делают обратную расчетную
операцию Сх=Ах/εl
. Ограничения:
обязательное подчинение закону
светопоглощения для аналитической
системы, т.е. концентрация анализируемого
раствора должна попасть во внутрь
диапазона концентрации раствора.М
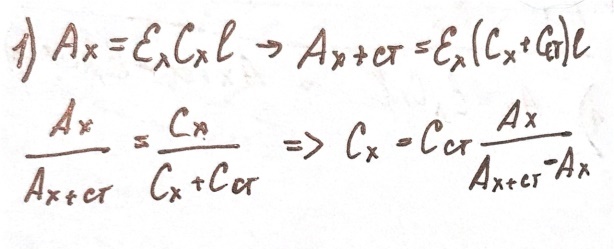 етод
добавок: применяют
при анализе растворов сложного состава
и очень разбавленных растворов. Суть:
определяют оптическую плотность Ах
анализируемого раствора, содержащего
определяемый компонент неизвестной
концентрации сх,
а затем в анализируемый раствор добавляют
известное количество определяемого
компонента (сст)
и вновь измеряют оптическую плотность
Ах+ст.
етод
добавок: применяют
при анализе растворов сложного состава
и очень разбавленных растворов. Суть:
определяют оптическую плотность Ах
анализируемого раствора, содержащего
определяемый компонент неизвестной
концентрации сх,
а затем в анализируемый раствор добавляют
известное количество определяемого
компонента (сст)
и вновь измеряют оптическую плотность
Ах+ст.

Метод дифференциальной фотометрии: метод применяют для повышения точности определения высоких концентраций в растворе. В качестве раствора сравнения используют один из стандартных растворов определяемого вещества. Точность будет тем выше ,чем ближе концентрация к концентрации вещества раствора сравнения.
Определение смеси светопоглощенных веществ.Метод Фирорта.Спектрофотометрический метод позволяет определить несколько светопоглощенных веществ в одном растворе без предварительного распределения. Имеем 2 вещества A и B ,вещества разные, длина волны-разные. В соответствии с законом аддитивности светопоглощения можно записать след. уравнения
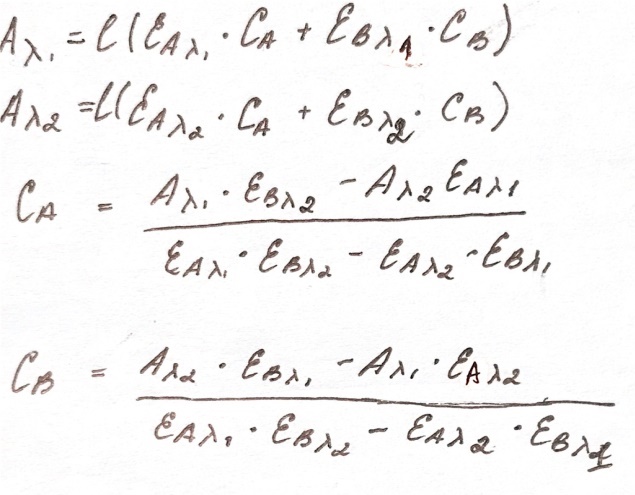
λ1 и λ2 , при которых идут А ,подбирают по спектропоглощениям А и В ,используют метод максимальной разности. Для этого делают серию стандартных А и стандартных растворов В.
Теоретические основы ИК-спектроскопии. Колебательные спектры.
ИК-спектроскопия - спектральный метод анализа, в основе которого лежит взаимодействие инфракрасного излучения с веществом.Свет поглощается веществом избирательно: при некоторых длинах волн светопоглощение происходит интенсивно, а при некоторых - свет не поглощается. Интенсивно поглощаются кванты света, энергия которых hν равна энергии возбуждения частицы, вероятность поглощения таких квантов больше 0. Распределение по частотам (или по длинам волн) значений молярного коэффициента (оптических плотностей, интенсивности света, прошедшего через поглощающую среду) называется спектром поглощения. Наибольший интерес представляют характеристики спектра: число полос поглощения, их положение по шкале длин волн, высота максимумов, форма полос поглощения.Появление полос поглощения обусловлено дискретностью энергетических состояний поглощающих частиц и квантовой природой электромагнитного излучения. Интенсивно поглощаются кванты света, которые соответствуют энергии возбуждения частицы. При поглощении квантов света происходит увеличение внутренней энергии частицы, которая складывается из энергии вращения частицы как целого, энергии колебания атомов в молекуле и энергии движения электронов: Е = Евр. + Екол. + Еэл.
Колебательные спектры
Полосы, связанные с возбуждением колебательных уровней энергии, расположены в области ИК спектра, что соответствует энергии квантов от 3 до 60 кДж/моль. Поэтому при обычных температурах энергетическое состояние молекул, как правило, характеризуется основным колебательным уровнем. Простейшей моделью, которая используется при рассмотрении колебаний двухатомной молекулы, является модель гармонического осциллятора. Кривая потенциальной энергии гармонического осциллятора обычно аппроксимируется параболой. Энергия может быть найдена по уравнению: 𝐸кол=(𝑉+12⁄)ℎ𝜈0, где V – колебательное квантовое число.
Сложную картину колебаний в многоатомной молекуле обычно представляют как суперпозицию нормальных колебаний.
По классификации нормальные колебания различают валентные и невалентные (деформационные) колебания. Колебания называют валентным, если происходит изменение длины связи без существенного изменения углов между связями. Колебания с изменением углов между связями называют деформационными.
Колебательный спектр используют для изучения строения молекул и для проведения качественного анализа в области ИК-спектра.
ИК- спектры. Характеристические частоты функциональных групп. Качественный анализ.
Обычный ИК-спектрометр работает по двухлучевой схеме: два параллельных световых потока пропускают через кювету с анализируемым образцом и через кювету сравнения. С помощью системы зеркал потоки поочередно попадают на монохроматор. В таких спектрометрах спектр регистрируется последовательно.Спектрометры с Фурье-преобразованием позволяют сразу получить информацию о спектре в форме интерферограммы. Путем Фурье-преобразования из интерферограммы можно получить спектр источника излучения.
Экспериментальные исследования большого числа молекул, обладающих одними и теми же химическими группами, показали, что, независимо от изменений в остальной части молекулы, эти одинаковые группы поглощают в узком интервале частот. Такие частоты получили название характеристические или групповые. Наличие групповых частот вызвано тем, что в таком колебании наибольшее участие принимает некоторая группа атомов, вклад остальной части молекулы мал, хотя, строго говоря, в каждом колебании изменяются длины всех связей и величины углов между связями.В двухатомной молекуле частота колебаний зависит от двух параметров: массы атомов и силового коэффициента, связанного с упругостью связи. В многоатомных молекулах дело обстоит намного сложнее, но тем не менее эта зависимость помогает очень грубо оценить области, в которых проявляются отдельные колебания и их характеристичность. Колебание будет тем более характеристичным, чем больше параметры колеблющейся группы атомов отличаются от параметров остальной части молекулы.
Факторы, влияющие на характеристические частоты
1) Агрегатное состояние, так как при его изменении изменяются межмолекулярные взаимодействия.
2) Водородные связи. С помощью ИК-спектроскопии можно различать внутри- и межмолекулярные водородные связи.
3) Электронные эффекты наблюдаются при введении в молекулу электронодонорных и электроноакцепторных заместителей.
4) Эффект массы атома. При замещении атома его более тяжелым изотопом приводит к сдвигу полосы в сторону меньших волновых чисел.
5) Влияние напряжения в циклах проявляется в том, что кратные связи в циклических структурах искажены тем сильнее, чем меньше размер цикла. Искажение кратных связей приводит к уменьшению силовых постоянных и частот колебаний.
6) Конформационные эффекты обусловлены различным взаимным расположением атомов и атомных групп в различных конформациях молекулы.
Качественный анализ методом ИК-спектроскопии – это идентификация веществ и расшифровка структуры веществ.
ИК-спектры можно использовать для:
установления природы вещества; для этого экспериментальный спектр неизвестного вещества сравнивают со спектрами, имеющимися в спектральной библиотеке;
для установления или исследования структуры вещества.
Для расшифровки молекулярной структуры можно использовать различные таблицы положений характеристических частот. Классические таблицы – таблицы Колтупа. Следует иметь в виду, что ни один метод (в том числе и ИК-спектроскопия) не может дать исчерпывающую информацию о структуре вещества, поэтому лучше использовать сочетание методов. В первую очередь определяют, к какому классу соединений относится исследуемое вещество, затем изучают его функциональный состав.
В какой области спектра находятся полосы поглощения, связанные с возбуждением колебательных уровней энергии?
Полосы, связанные с возбуждением колебательных уровней энергии, расположены в области спектра примерно от 200...300 до 4000...5000 см-1, что соответствует энергии квантов от 3 до 60 кДж/моль. Поэтому при обычных температурах энергетическое состояние молекул, как правило, характеризуется основным колебательным уровнем. Простейшей моделью, которая используется при рассмотрении колебаний двухатомной молекулы, является модель гармонического осциллятора. Это система из двух масс, связанных упругой силой. Кривая потенциальной энергии гармонического осциллятора обычно аппроксимируется параболой (рис. 3, кривая 1).
Следует отметить лишь, что колебательными инфракрасными спектрами обладают не все молекулы, а только те, у которых при колебании происходит изменение их электрического дипольного момента.
ИК-спектрами обладают, например, молекулы НС1, НВг и т. д., но не Н2, О2 и др.
Колебательные спектры многоатомных молекул интерпретируют на основе учения о симметрии молекул и теории групп. Математический аппарат теории групп позволяет вычислить число частот и правила отбора для молекул различной симметрии ( для определения молекулярных констант, изучения строения молекул). Для решения химико-аналитических задач используются так называемые характеристические частоты(чаще для качественного анализа). Анализ ИК-спектров показал, что некоторые из наблюдаемых частот можно привести в соответствие с колебаниями отдельных атомов или групп атомов. Так, например, было найдено, что в спектрах всех молекул, содержащих связи С-Н, имеются частоты в области 2800...3000 см-1, тройная связь С-С характеризуется частотой 1650 см-1, а тройная связь C-C- частотой 2100 см-1
Какова энергия квантов света, вызывающих колебательные переходы?
Энергия квантов света, вызывающих колебательные переходы, равна от 3 до 60 кДж/моль.
Энергию квантов в физике принято выражать в электрон-вольтах. Это внесистемная единица измерения энергии. Один электрон-вольт (1 эВ) равен энергии, которую приобретает электрон, когда разгоняется электрическим полем напряжением 1 вольт. Это очень небольшая величина, в единицах системы Си 1 эВ = 1,6·10–19 Дж. Но в масштабах атомов и молекул электрон-вольт — вполне солидная величина.
От энергии квантов напрямую зависит способность излучения производить определенное воздействие на вещество. Многие процессы в веществе характеризуются пороговой энергией — если отдельные кванты несут меньшую энергию, то, как бы много их ни было, они не смогут спровоцировать надпороговый процесс.
Немного забегая вперед, приведем примеры. Энергии СВЧ-квантов хватает для возбуждения вращательных уровней основного электронно-колебательного состояния некоторых молекул, например воды. Энергии в доли электрон-вольта хватает для возбуждения колебательных уровней основного состояния в атомах и молекулах. Этим определяется, например, поглощение инфракрасного излучения в атмосфере. Кванты видимого света имеют энергию 2–3 эВ — этого достаточно для нарушения химических связей и провоцирования некоторых химических реакций, например, тех, что протекают в фотопленке и в сетчатке глаза. Ультрафиолетовые кванты могут разрушать более сильные химические связи, а также ионизировать атомы, отрывая внешние
электроны. Это делает ультрафиолет опасным для жизни. Рентгеновское излучение может вырывать из атомов электроны с внутренних оболочек, а также возбуждать колебания внутри атомных ядер. Гамма-излучение способно разрушать атомные ядра, а самые энергичные гамма-кванты даже внедряются в структуру элементарных частиц, таких как протоны и нейтроны.
Полосы, связанные с возбуждением колебательных уровней энергии, расположены в области спектра примерно от 200...300 до 4000...5000 см-1, что соответствует энергии квантов от 3 до 60 кДж/моль. Поэтому при обычных температурах энергетическое состояние молекул, как правило, характеризуется основным колебательным уровнем. Простейшей моделью, которая используется при рассмотрении колебаний двухатомной молекулы, является модель гармонического осциллятора.
Что из себя представляют колебательные спектры? Каковы их особенности?
Полосы, связанные с возбуждением колебательных уровней энергии, расположены в области ИК спектра, что соответствует энергии квантов от 3 до 60 кДж/моль. Простейшей моделью, которая используется при рассмотрении колебаний двухатомной молекулы, является модель гармонического осциллятора. Кривая потенциальной энергии гармонического осциллятора обычно аппроксимируется параболой. Энергия может быть найдена по уравнению: 𝐸кол=(𝑉+12⁄)ℎ𝜈0, где V – колебательное квантовое число.
Сложную картину колебаний в многоатомной молекуле обычно представляют как суперпозицию нормальных колебаний.
Совокупность серий полос, отвечающих переходу молекулы с данного колебательного уровня на соседние, представляет собой колебательный спектр. Если переходы сопровождаются поглощением энергии, то спектр называется спектром поглощения. Спектр поглощения можно получить, если на пути электромагнитного излучения помещено вещество, поглощающее лучи определенных длин волн.
Колебательные уровни молекул расположены на сравнительно близких расстояниях друг от друга так, что частота ν колебательного спектра относится к инфракрасной области спектра с длиной волны в несколько микрон.
Колебательные спектры многоатомных молекул отличаются высокой специфичностью и представляют сложную картину, хотя общее число экспериментально наблюдаемых полос могут быть существенно меньше возможного их числа, теоретически отвечающего предсказываемому набору уровней. Обычно, основным частотам соответствуют более интенсивные полосы в колебательном спектре. Некоторые из полос колебательного спектра могут наблюдаться только в инфракрасном или только в спектре комбинационного рассеяния, другие - с разной интенсивностью в обоих спектрах, а некоторые вообще экспериментально не наблюдаются.
По классификации нормальные колебания различают валентные и невалентные (деформационные) колебания. Колебания называют валентным, если происходит изменение длины связи без существенного изменения углов между связями. Колебания с изменением углов между связями называют деформационными.
Колебательный спектр используют для изучения строения молекул и для проведения качественного анализа в области ИК-спектра.
Колебания каких молекул можно описать кривой потенциальной энергии ангармонического осциллятора?
Ангармонические колебания можно отличить от гармонических уже по тому, что принцип суперпозиции не действует: сумма понуждающих колебание сил не единственное, что влияет на осциллятор. Системы, порождающие такие колебания, называются нелинейными. Нелинейные колебательные движения по сути – это когда к основному колебанию примешиваются еще другие гармоники – тоже колебания, которые в сумме создают неравномерные, изменяющиеся во времени колебания.
В действительности любая молекула – это ангармонический осциллятор. На рис.3 представлена кривая потенциальной энергии ангармонического осциллятора.
Энергетические уровни ангармонического осциллятора с увеличением квантового числа сближаются. Их положения ограничены энергией диссоциации молекулы. Разрешены переходы между любыми уровнями, следовательно, в спектре наблюдается несколько полос. Наиболее интенсивной является первая полоса, возникающая при переходе с уровня V=0 на уровень V =1. Этой полосе соответствует основная или фундаментальная частота. Менее интенсивные полосы дают обертоны, т.е. частоты, характеризующие переход с уровня V=0 на уровень V =2 (первый обертон или вторая гармоника), на уровень V =3 (второй обертон или третья гармоника).
Колебательными или ИК-спектрами обладают не все молекулы, а только те, у которых при колебании происходит изменение их электронного дипольного момента, т.е. полярные молекулы (Напимер, HCl, HBr). Неполярные молекулы (О2, Н2 и др.) не обладают ИК-спектрами.
Сравнительная простота колебательных спектров двухатомных молекул обусловлена тем, что колебания происходят только вдоль линии, соединяющей ядра. В многоатомной молекуле колеблются все атомы. Число колебательных степеней свободы у нелинейной молекулы, состоящей из N атомов равно: f = 3N – 6, а у линейной f = 3N – 5.
Какие молекулы обладают ик-спектрами? у каких молекул нельзя наблюдать ик-спектры?
ИК-спектрами обладают те молекулы, у которых при колебании происходит изменение их электронного дипольного момента. К ним относятся полярные молекулы. ИК-спектры нельзя наблюдать у неполярных молекул.
В ИК спектрах многоатомных молекул проявляются те колебания, которые происходят с изменением дипольного момента.
Что такое нормальные колебания молекулы? Рассмотреть виды нормальных колебаний на примере молекулы воды.
Норма́льные колеба́ния, со́бственные колебания или мо́ды — набор характерных для колебательной системы типов гармонических колебаний. Каждое из нормальных колебаний физической системы, например, колебаний атомов в молекулах, характеризуется своей частотой. Такая частота называется нормальной частотой, или собственной частотой (по аналогии с линейной алгеброй: собственное число и собственный вектор). Набор частот нормальных колебаний составляет колебательный спектр.
Виды нормального колебания:
- валентные: представляют собой растяжение или сжатие связей атомов в молекуле; углы между связями остаются примерно неизменными. Существуют симметричные и антисимметричные колебания.
- деформационные (невалентные): это типы колебаний, которые сопровождаются изменением углов между связями молекул. Такие колебания связываются с более низкими величинами силовых 12 постоянных; соответствующие полосы поглощения появляются при более низких частотах по сравнению с валентными.
У воды два типа валентных колебаний и один тип деформационный.
Симметричное: атомы водорода колеблются в направлении по связи с атомом кислорода, кислород же – в направлении от них и к ним.
Несимметричное: атомы водорода колеблются по связи друг от друга, атом кислорода – в противоположном направлении.
Деформационное: атомы водорода сближаются и отдаляются, меняя угол между связями, атом кислорода колеблется зависимо от них.
f = 3∙3 – 6 = 3 – число нормальных степеней свободы. У молекулы воды 2 типа валентных колебаний и один тип деформационных.
Валентные колебания

 v
1
v
1











v 2











Невалентные (деформационные) колебания δ










Рис.4. Нормальные колебания молекулы воды.
Как устроен ИК-спектрометр? Кратко описать основные узлы и принцип его работы.
Устройство традиционного ИК-спектрометра
Инфракрасные спектрометры с Фурье-преобразованием (ИКФС) предназначены для регистрации спектров пропускания, поглощения и разных видов отражения твёрдых, жидких, порошковых, пастообразных, газообразных образцов в инфракрасной области электромагнитного спектра. Применимость таких спектрометров в настоящее время охватывает практически все сферы человеческой деятельности, связанные с определением молекулярного строения и молекулярного состава, например, с определением подлинности, чистоты, качества и стабильности веществ, их точной идентификацией как в индивидуальном виде, так и в составе смесей.
2.1.1. Источник излучения
В этом качестве используют раскаленные твердые тела. Распределение интенсивности излучения по длинам волн зависит от температуры. Для ИК-спектроскопии используют более длинноволновое (чем в видимой области) и относительно менее интенсивное излучение в области 4000 – 400(200) см-1.
Наиболее распространены в качестве источника возбуждения:
штифт Нернста - стержень из оксидов иттрия и циркония, рабочая температура 1900 0С;
глобар - из карбида кремния (SiC), рабочая температура 1350 0С;
менее интенсивные, но более продолжительные в эксплуатации источники излучения из тугоплавких сплавов (хрома, никеля);
ртутные разрядные лампы высокого давления для дальней ИК-области 200 –10 см-1
обычные вольфрамовые лампы накаливания для ближней инфракрасной области 4000 – 12800 см-1, 2500 -780 нм).
2.1.2. Отделение для проб
В ИК-спектроскопии пробоподготовка более трудоемка. Используются кюветы из галогенидов щелочных металлов.
Газообразные пробы. Используют специальные вакуумированные кюветы, толщиной от нескольких миллиметров до многих метров.
Жидкие пробы. Вода и спирт в качестве растворителей не пригодны, поскольку интенсивно поглощают в ИК- области, а также взаимодействуют с материалом кювет. Любой органический растворитель необходимо тщательно избавлять от следов воды. Выбор растворителя, не поглощающего ИК-излучение, представляет серьезную проблему. Чаще всего используют ацетон, сероуглерод, четыреххлористый углерод (CCl4), хлороформ, диэтиловый эфир, циклогексан, бензол. Чтобы собственное поглощение растворителя было как можно меньше, используют кюветы с толщиной от 0,01 до 1 мм.
Твердые образцы можно исследовать непосредственно, если из материала образца можно приготовить тонкий слой. Как правило, на специальном гидравлическом прессе готовят таблетку из тонко растертой смеси, состоящей из оптически чистого KBr и материала образца в соотношении 100 мг: несколько мг. Для спектроскопии в дальней ИК-области используют полиэтилен.
Вместо KBr может быть использован нуйол – длинноцепочечная парафиновая фракция нефти, представляющая собой вязкую жидкость. Смешивают измельченную в порошок пробу с нуйолом – получают пастообразную суспензию. Помещают это между двумя окошками кюветы и плотно зажимают. В состав нуйола входят группы – СН2, – СН3, имеющие собственное поглощение в ИК-области, поэтому определение этих структурных фрагментов в образце невозможно. Образцы с бромидом калия очень чувствительны к атмосферному воздуху и влаге. Используя нуйол, можно этого не опасаться. Недостатком нуйола является невозможность анализа водных растворов биологических жидкостей (сильнО поглощение растворителя).
2.1.3. Монохроматоры. Применяют призмы из кварца, LiF, NaCl, KBr, CsF в зависимости от исследуемого спектрального диапазона, а также дифракционные решетки.
2.1.4. Детекторы – термопары и балометры. Термопара преобразует энергию ИК-излучения в тепловую, а затем в электрическую. Балометр работает по принципу термометра сопротивления.
Общая проблема при измерении интенсивности ИК-излучения - наличие значительного теплового шума окружающей среды.
Обычный ИК-спектрометр работает по двухлучевой схеме: два параллельных световых потока пропускают через кювету с анализируемым образцом и через кювету сравнения. С помощью системы зеркал потоки поочередно попадают на монохроматор. В таких спектрометрах спектр регистрируется последовательно.
Спектрометры с Фурье-преобразованием позволяют сразу получить информацию о спектре в форме интерферограммы. Путем Фурье-преобразования из интерферограммы можно получить спектр источника излучения.
В чем заключаются особенности и трудности в пробоподготовке для проведения анализа методом ИК-спектроскопии?
В ИК-спектроскопии пробоподготовка более трудоемка. Используются кюветы из галогенидов щелочных металлов.
Газообразные пробы. Используют специальные вакуумированные кюветы, толщиной от нескольких миллиметров до многих метров.
Жидкие пробы. Вода и спирт в качестве растворителей не пригодны, поскольку интенсивно поглощают в ИК- области, а также взаимодействуют с материалом кювет. Любой органический растворитель необходимо тщательно избавлять от следов воды. Выбор растворителя, не поглощающего ИК-излучение, представляет серьезную проблему. Чаще всего используют ацетон, сероуглерод, четыреххлористый углерод (CCl4), хлороформ, диэтиловый эфир, циклогексан, бензол. Чтобы собственное поглощение растворителя было как можно меньше, используют кюветы с толщиной от 0,01 до 1 мм.
Твердые образцы можно исследовать непосредственно, если из материала образца можно приготовить тонкий слой. Как правило, на специальном гидравлическом прессе готовят таблетку из тонко растертой смеси, состоящей из оптически чистого KBr и материала образца в соотношении 100 мг: несколько мг. Для спектроскопии в дальней ИК-области используют полиэтилен.
Вместо KBr может быть использован нуйол – длинноцепочечная парафиновая фракция нефти, представляющая собой вязкую жидкость. Смешивают измельченную в порошок пробу с нуйолом – получают пастообразную суспензию. Помещают это между двумя окошками кюветы и плотно зажимают. В состав нуйола входят группы – СН2, – СН3, имеющие собственное поглощение в ИК-области, поэтому определение этих структурных фрагментов в образце невозможно. Образцы с бромидом калия очень чувствительны к атмосферному воздуху и влаге. Используя нуйол, можно этого не опасаться. Недостатком нуйола является невозможность анализа водных растворов биологических жидкостей (сильнО поглощение растворителя).
Что такое характеристические частоты?
Экспериментальные исследования большого числа молекул, обладающих одними и теми же химическими группами, показали, что, независимо от изменений в остальной части молекулы, эти одинаковые группы поглощают в узком интервале частот. Такие частоты получили название характеристических или групповых.
Существование характеристических частот объяснено следующим образом. Колебания определенной группы атомов или связей могут быть слабо связаны с колебаниями атомов остальной части молекулы. В этом случае частота колебаний этой группы или связи зависит только от их строения и мало зависит от окружающих атомов и связей. Вследствие этого различные молекулы, содержащие данную группу атомов или связей, будут характеризоваться различными колебательными спектрами, однако в каждом из них будет присутствовать одна или несколько одинаковых или почти одинаковых частот. Установление характеристических частот позволяет, не производя никаких расчетов, определять по спектру присутствие в молекуле различных групп и связей и тем самым установить строение молекулы.
Некоторые колебания могут быть локализованы главным образом на некоторых атомах и не затрагивать остальную часть молекулы. Это наблюдается в тех случаях, когда частоты колебаний группы атомов и остального остова сильно различаются между собой. А это, в свою очередь, имеет место, когда в рассматриваемый структурный фрагмент входят либо атомы, значительно отличающиеся от остальных по своей массе, либо связи со значительно отличающейся силовой постоянной.
Наличие таких частот колебаний позволяет делать вывод о строении изучаемых молекул и имеет большое значение для молекулярного спектрального анализа. По инфракрасным спектрам вещество может быть идентифицировано. Можно определить симметрию и структуру молекул, термодинамические характеристики; провести количественный анализ, изучить химические равновесия и кинетику химических реакций, контролировать ход технологических процессов.
Какие факторы влияют на характеристические часиоты?
На характеристические частоты влияют: 1. Агрегатное состояние, так как при его изменении изменяются межмолекулярные взаимодействия.
2. Водородные связи. С помощью ИК-спектроскопии можно различать внутри- и межмолекулярные водородные связи.
Чем прочнее водородная связь, тем ниже частота колебаний. Зависимость частоты колебаний ОН-связи от концентрации наблюдается только в случае межмолекулярных водородных связей, т.к. при разбавлении уменьшается степень ассоциации молекул.
3. Электронные эффекты наблюдаются при введении в молекулу электронодонорных и электроноакцепторных заместителей.
4. Эффект массы атома. При замещении атома его более тяжелым изотопом приводит к сдвигу полосы в сторону меньших волновых чисел.
5. Влияние напряжения в циклах проявляется в том, что кратные связи в циклических структурах искажены тем сильнее, чем меньше размер цикла. Искажение кратных связей приводит к уменьшению силовых постоянных и частот колебаний.
6. Конформационные эффекты обусловлены различным взаимным расположением атомов и атомных групп в различных конформациях молекулы.
Как проявляются взаимодействия колебаний многоатомной молекулы?
Сравнительная простота колебательных спектров двухатомных молекул обусловлена тем, что колебания происходят только вдоль линии, соединяющей ядра. В многоатомной молекуле колеблются все атомы. Число колебательных степеней свободы у нелинейной молекулы, состоящей из N атомов равно: f = 3N – 6, а у линейной f = 3N – 5.
Принято называть колебания в многоатомной молекуле нормальными колебаниями. Частота нормальных колебаний характеризуется положением пол
Число нормальных колебаний равно числу колебательных степеней свободы.
Нормальные колебания подразделяются на:
валентные и
невалентные (деформационные).
Валентные колебания - это колебания, в результате которых происходит изменение длины связи без существенного изменения углов между связями. Обозначают их v.
Невалентные (деформационные) происходят с изменением угла связи, обозначают их δ.
Пример. Нормальные колебания молекулы воды H2O.
f = 3∙3 – 6 = 3 – число нормальных степеней свободы. У молекулы воды 2 типа валентных колебаний и один тип деформационных.
Колебание многоатомных молекул
Основные типы колебаний молекулы называются нормальными колебаниями. Например, для молекулы воды имеются три возможности колебательных движений: два валентных (аи б) и одно деформационное (в)











а) νs б) νаs в) δ
Более строго, нормальные колебания – это колебания, которые происходят независимо друг от друга, т.е. при возбуждении нормального колебания не происходит никакой передачи энергии для возбуждения других колебаний.
С точки зрения формы колебаний различают: валентные колебания ν, которые происходят в направлении химической связи, и при которых изменяется межатомное расстояние; деформационные колебания (невалентные) δ – изменяются валентные углы, межатомные расстояния остаются постоянными.
Каково основное назначение ИК-спектроскопии? (Область применения).
Метод инфракрасной спектроскопии дает возможность с высокой вероятностью предсказывать качественный количественный состав химических соединений. Современные приборы позволяют осуществлять процедуру измерения этих показателей с достаточной точностью и высокой воспроизводимостью результатов измерений.
ИК - спектроскопия находит применение в исследовании строения полупроводниковых материалов, полимеров, биологических объектов и живых клеток. Также она играет большую роль в создании и изучении молекулярных оптических квантовых генераторов, излучение которых лежит в инфракрасной области спектра.
Химия и нефтехимия. Качественный и количественный анализ сырья, промежуточных и конечных продуктов синтеза. Фракционный и структурно-групповой состав нефтепродуктов. Анализ топлив: эфиры, спирты, ароматика, октановое число.
Химия полимеров. Анализ сополимеров. Синтетические каучуки: состав, структурные характеристики. Анализ модифицирующих добавок: пластификаторы, антиоксиданты.
Фармацевтическая промышленность. Определение подлинности субстанций по ИК-стандартам, контроль качества лекарственных форм и сырья.
Газовый анализ. Анализ многокомпонентных газовых смесей. Контроль качества продукции газовой промышленности, анализ состава и влажности природного газа.
Криминалистический, судебно-медицинский и биоклинический анализ. Качественный и количественный анализ природных веществ и продуктов синтеза. Идентификация наркотиков, ОВ и ВВ. Анализ следовых остатков веществ.
Области ИК-спектроскопии наиболее частого применения:
1. Исследование строения, установление функциональных групп по найденным характеристическим частотам.
2. Установление идентичности, например, исследуемого вещества с известным образцом, которые должны иметь тождественные спектры (как в области характеристических частот, так и в области скелетных колебаний).
3. Решение задач. Определение чистоты: наличие «посторонних» пиков свидетельствует о примесях.
4. Количественный анализ: интенсивность поглощения в определенных частях спектра пропорциональна концентрации вещества.
5. Изучение внутри- и межмолекулярных взаимодействий, например установление наличия водородных связей.
Каковы основные направления качественного анализа?
Задачей качественного анализа является идентификация компонентов и определение качественного состава вещества или смеси веществ.
Для этого используют химические реакции с характерным внешним эффектом (выделением газа, появлением, исчезновением или изменением осадка или окраски и т.п.), а также физические свойства веществ (температуры плавления или кипения, коэффициенты преломления, положение полос поглощения, испускания или люминесценции в спектре и т.д.).
Обнаружение или, как говорят, открытие элементов или ионов в составе исследуемого вещества производят, переводя их в соединение, обладающее какими-либо характерными свойствами, т. е. фиксируют появление аналитического сигнала. Происходящие при этом химические превращения называются аналитической реакцией.
Качественный анализ методом ИК-спектроскопии используется для идентификации веществ и расшифровки структуры веществ. Спектры поглощения используются для изучения химического равновесия, кинетики химических реакций, строения вещества, для изучения взаимодействия между частицами в растворах и т.д. Используются такие направления, как:
Полумикроананализ (полумикрометод) - в данном методе сохраняется система макроанализа и открытия ионов, но все реакции выполняют с малыми количествами вещества, пользуясь специальной техникой и аппаратурой.
Субмикроанализ и ультрамикроанализ проводятся по специальным методикам с использованием микроскопов разной степени увеличения, электронных микроскопов и другой аппаратуры. Примером таких реакций могут служить реакции окрашивания пламени солями некоторых металлов.
Каковы возможности количественного анализа методом ИК-спектроскопии?
Спектральная область 4000 – 12500 см-1 (2500 -800 нм) называется ближней ИК-областью; 4000-400 - средней.
Средняя область пригодна для количественного анализа гораздо хуже, чем УФ и ВД (видимая).
Причины следующие.
Интенсивность источников и детекторов невелика.
Недостаточная монохроматичность излучения, и как следствие, зависимость оптической плотности от концентрации плохо (или не подчиняется) подчиняется закону Бугера-Ламберта-Бера.
Уровень рассеянного излучения выше.
Пути решения проблемы.
Тщательная градуировка с использованием стандартных образцов, применения современной аппаратуры, опыт исследователя.
Более пригодна для анализа ближня область ИК-спектра.
Метод инфракрасной спектроскопии может быть также применен для количественного анализа минералов смеси. В соответствии с законом Бугера-Ламберта-Бера, интенсивность полос спектра поглощения, помимо других характеристик образца вещества зависит от соотношения концентраций компонентов веществ (принимается ли во внимание механическая смесь минералов или «смесь» на основе изоморфного замещения).
В соответствии с этим законом количественный анализ основан на измерении интенсивности полос спектра поглощения. С большей или меньшей точностью определение содержания компонентов минералов производится по методикам, которые первоначально предусматривают построение графиков зависимости интенсивности полос поглощения каждого компонента от его содержания в смеси.
Определение одного вещества методом ИК-спектроскопии базируется на основном законе светопоглощения. Его используют для количественного определения органических веществ. Анализ смесей же веществ базируется не только на законе светопоглощения, но и на законе аддитивности оптических плотностей.
Рефрактометрический метод анализа. Теоретические основы. Область применения метода.
Рефракция- это явление преломления света на границе раздела двух сред, различных по оптической плотности. Рефрактометрия-это измерение преломления света. Количественно рефракцию оценивают по углу или показателю преломления света, поэтому рефрактометрический метод анализа -это метод, основанный на зависимости угла или показателя преломления света от состава системы (т.к. каждая система отличается определенной оптической плотностью). Чаще всего для количественной оценки преломления света используют показатель преломления.
П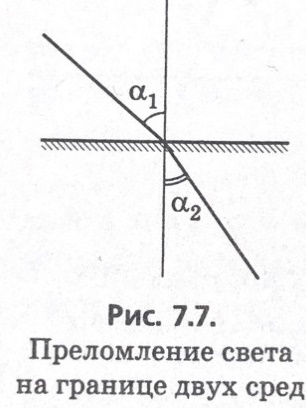 ервая
среда менее плотная. Мы должны добиваться
максимального угла преломления.
ервая
среда менее плотная. Мы должны добиваться
максимального угла преломления.
Показатель преломления: различают 2 типа(абсолютная и относительная).
Абсолютный(N) показатель отношение Vвак/Vсреды
Относительный(n) n=Vсреды1/Vсреды2 n=Vвозд/Vсреды
Закон преломления света: n21=sinα/sinβ ; n2/n1= sinα/sinβ=1/sinβmax; n1=n2sinβmax Показатель преломления всегда величина больше 1.
Зависит о различных факторов:
От природы вещества-каждое вещество состоит в оптимальном состоянии, состоит из определённых частиц, молекул и ионов, которые препятствуют прохождения света . Показатель свой ,индивидуальный.
Температура –с увеличением, показателя преломления уменьшается
Длина волны; дисперсия (зависимость от длины волны), все табличные знчения,даны относительно желтой линии (D).Разность показателя преломления называется частотой дисперсии.
Концетрация(для газов давление):чем больше частичек, чем больше показатель
Поляризация и рефракция
При прохождении э/м волны через вещества происходит смещение положительных и отрицательных зарядов в молекулах. Образуется диполь с наведенным моментом 𝜇=𝑒𝑙
Именно это явление называется поляризацией.
При поляризации молекулы ориентируются вдоль э/м поля.
Это явление называется поляризация-ориентация (Ро)
Когда молекула не была полярной, поляризация-деформация (Рд)
Поляризация-деформация складывается из смещения электронов и атомов. Рд=Ре+Ра Р=Ре+Ра+Ро
Рд в расчете на моль вещества определяется: Рд=43𝜋𝛼𝑁𝐴
Альфа связана с диэлектрической проницаемостью среды 43𝜋𝛼𝑁𝐴=[(𝜀−1)(𝜀+1)⁄]𝑀𝜌 Рд=(𝜀−1𝜀+1)𝑀𝜌=(𝑛2−1𝑛2+1)𝑀𝜌
𝑅𝑛=(𝑛2−1𝑛2+1)𝑀𝜌 – уравнение Лоренца-Лоренца
Молярная рефракция определяется только поляризуемостью молекул, поэтому зависит только от природы вещества, следовательно по Rn можно производить идентификацию вещества. Рефракция – мера поляризуемости молекулы.
Рефракция – это мера поляризуемости молекул, которая складывается из поляризуемости атомов, составляющих молекулы. Следовательно, рефракция есть величина аддитивная и может быть получена как сумма рефракцией атомов с учётом их валентного состояния и особенности расположения.
Практическое применение и достоинства.
Достоинства: простота прибора, точность, малое количество вещества.
Применение: качественные и количественные анализ, структурная формула вещества, определить степень частоты вещества, установление структурной формулы молекулы.
Качественный анализ основан на зависимости показателя преломления от природы вещества.
Различные способы классификации электрохимических методов анализа.
Электрохимические методы подразделяют на равновесные (потенциометрию) и неравновесные (кондуктометрию, вольтамперометрию, электрогравиметрию и кулонометрию).
В случае равновесных методов аналитическим сигналом является электродный потенциал индикаторного электрода, измеряемый в равновесных условиях, когда сила тока в электрохимической цепи практически равна нулю. Индикаторным в этом случае выбирают электрод, потенциал которого обратимо изменяется при изменении активности (концентрации) определяемого вещества в контактирующем с электродом растворе.
Если же через ячейку протекает электрический ток (происходит электролиз и реализуются неравновесные условия), то для химического анализа можно использовать зависимость силы тока от напряжения. Эти м-ды называются вольтамперометрическими. Методы, основанные на измерении напряжения при постоянной силе тока, называют вольтамперометрическими, а на измерении силы тока при постоянном напряжении – амперометрическими. вольтамперометрический анализ проводят в условиях, когда степень электрохимического превращения определяемого вещества вследствие электролиза пренебрежимо мала. Однако электролиз можно проводить и до полного превращения определяемого вещества. На этом основаны неравновесные методы электрогравиметрии и кулонометрии. К неравновесным методам относится и кондуктометрия, в которой аналитическим сигналом служит электропроводность раствора электролита, являющаяся функцией его концентрации.
Все электрохимические методы можно разделить на прямые и косвенные. Косвенные- методы титрования, в которых конечную точку определяют электрохимическим способом.
Механизмы переноса: конвекцию, миграцию и диффузию. Конвекцией называется перенос вещества макроскопическими потоками. Миграция – это движение заряженных частиц под действием электрического поля. Диффузия – это процесс переноса вещества под действием разности химических потенциалов.
5 методов: 1) потенциометрия – без положения внешнего пот-ла ( Прямая и косвенная-ППТ). Аналит сигнал- пот-л эл-да
2) Кондуктометрия (прямая и косвенная). Ан.с.- электропроводность р-ра
3) Кулонометрия. Ан.с – кол-во эл-ва
4) Электрогравиметрич. Ан-з. А.с – изменение массы эл-да
5) вольтамперометрич.
Гальванический элемент. ЭДС гальванического элемента. Вывод уравнения Нернста.
Стандартный эл-нт – нормальный эл-нт Вестона, электродвиж сила кот-го обладает строго постоянным воспроизводимым значением, сохраняющимся в течении многих лет, и незначительным температурным коэф-том. Электродами являются ртуть и насыщенный амальгама кадмия – насыщенный водный р-р CdSO4*8/3H2O и Hg2SO4. Что бы обеспечить сущ-ие насыщ-го р-ра, в эл-нт в контакте с эл-литом вводят кристаллы этих соединений. Эл-хим цепь эл-нта Вестона имеет вид :
(–)Cd | CdSO4 || SO42-, Hg2SO4 | Hg (+)
Гальв-ий эл-нт , состоящий из водородного и хлорсер-го эл-дов условно изображен: Pt, (H2)| 0,1M H2SO4|| 0,1M KCl, Ag. Электродвиж сила такой цепи измеряется компенсационным методом, когда ЭДС исслед-го эл-нта точно компенсируется внешним источником напряжения и через элемент тока практически не проходит и в системе не протекают процессы, ведущие к заметным хим или концентрационнымизменениям за счет эл-за.
ГЭ преобразует химич. энергию в электрич. :ЕЭДС = ЕКАТОДА – ЕАНОДА
ЕЭДС > 0 для самопроизв. работы.
ГЭ состоит из:
индикаторного электрода, потенциал которого зависит от конц. (акт-ти) опред. иона,
электрода сравнения, потенциал которого должен оставаться постоянным независимо от протекания каких-либо р-ций анализир. р-ре.
ЭДС ГЭ – сумма скачков потенциала на всех границах.
Потенциал ЭХ ячейки предст-ет собой ГЭ.
Потенц. методы основаны на измерении ЭДС, которые пред-ют собой напряжение на электродах в отсутствии тока. В этом случае, один из электродов – неполяризуемый индикатор электрода, другой – электрод сравнения. Потенциал электрода сравнения связан с акт-тью и конц. вещ-в, участв-щих в процессе, ур-ем Нернста.
уравнение Нернста:
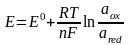 =
=
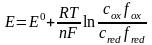 ,
,
где E0 – cтандартный потенциал редокс-системы; R – универсальная газовая постоянная; Т – абсолютная температура; F – постоянная Фарадея; n – число электронов, принимающих участие в электродной реакции; aox,, ared – активности окисленной и восстановленной форм редокс-системы; сox,, сred – их молярные концентрации; fox,, f red – коэффициенты активности.
Вывод уравнения Нернста
![]() ,
,
где
 —
электродный потенциал,
—
электродный потенциал,
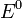 —
стандартный электродный потенциал,
измеряется в вольтах;
—
стандартный электродный потенциал,
измеряется в вольтах; —
универсальная газовая
постоянная, равная 8.31 Дж/(моль·K);
—
универсальная газовая
постоянная, равная 8.31 Дж/(моль·K); —
абсолютная температура;
—
абсолютная температура; —
постоянная Фарадея,
равная 96485,35 Кл·моль−1;
—
постоянная Фарадея,
равная 96485,35 Кл·моль−1;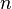 —
число моль электронов,
участвующих в процессе;
—
число моль электронов,
участвующих в процессе;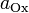 и
и
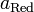 —
активности соответственно окисленной
и восстановленной форм вещества,
участвующего в полуреакции.
—
активности соответственно окисленной
и восстановленной форм вещества,
участвующего в полуреакции.
Если в формулу Нернста
подставить числовые значения констант
и
и
перейти от натуральных логарифмов к
десятичным, то при
![]() получим
получим
![]()
Стандартный электродный потенциал. Стандартный электрод сравнения.
Стандартный электродный потенциал – это электродный потенциал, измеренный при стандартных условиях относительно стандартного водородного электрода. Зависит только от температуры, давления и природы растворителя.
(активность ионов = 1, p = 1атм.)
Водородный электрод – электрод из платиновой пластинки, покрытый мелкодисперсной платиной, пластина погружена в раствор серной или соляной кислоты (1М). Через сосуд пропускают водород под давлением 1 атм., в этом случае платинированная платина поглощает газообразный водород и электрод действует так будто состоит из водорода, находящегося в равновесии со своими ионами, то есть 2H+ + 2e = H2↑. Потенциал стандартного водородного электрода 0 при любой t.
Стандартный электрод сравнения – потенциал которого должен оставаться постоянным независимо от протекания каких-либо реакций в анализируемом растворе.
уравнение Нернста:
= ,
где E0 – cтандартный потенциал редокс-системы; R – универсальная газовая постоянная; Т – абсолютная температура; F – постоянная Фарадея; n – число электронов, принимающих участие в электродной реакции; aox,, ared – активности окисленной и восстановленной форм редокс-системы; сox,, сred – их молярные концентрации; fox,, f red – коэффициенты активности.
Но потенциал отдельного электрода экспериментально определить невозможно (мы можем измерить только разницу потенциалов). Относительные значения электродного потенциала находят, комбинируя данный электрод со стандартным водородным электродом, который является общепринятым международным стандартом. Его потенциал принят равным нулю при всех температурах, поэтому потенциал данного электрода – это, в сущности, ЭДС элемента, состоящего из данного и стандартного водородного электродов. Водородный станд. электрод пред-ет собой платинированную платиновую пластинку, омываемую газообразным водородом при давлении р=1 и погруженную в р-р кис-ты с акт-тью аН+ = 1. При работе водород. эл-да протекает р-ция: Н2(г.) = 2Н+ + 2е-. На практике вместо хрупкого и капризного водородного эл-да исп-ют спец., более удобные в работе стабильные эл-ды сравн-я, потенциал которых по отношению к станд. водород. эл-ду точно известен. Например, AgCl эл-д (Е=+0,222 В).
Стандартные электроды. Вывести уравнение, описывающее потенциал хлорид-серебряного электрода.
В качестве электрода сравнения, потенциал которого принимается за нуль отсчета, по предложению В.Нернста принят стандартный водородный электрод. На этом электроде осуществляется равновесный окислительновосстановительный процесс: 2Н+ + 2е ⇔ 2Надс. ⇔ Н2
Стандартный водородный электрод представляет собой сосуд, заполненный кислотой (как правило, Н2SO4 ) в котором находится платиновая пластинка, служащая для адсорбции молекулярного водорода и его диссоциации на атомы, а также являющаяся проводником первого рода, поставляющим в систему свободные электроны.
Электроды второго рода состоят из металла, покрытого слоем малорастворимого соединения этого металла и погруженного в раствор хорошо растворимого соединения с тем же анионом. К ним относятся хлорсеребряный, каломельный и др. Обычно их применяют в качестве электродов сравнения.
Электроды сравнения должны обладать устойчивым во времени воспроизводимым потенциалом, не меняющимся при прохождении небольшого тока. Хлорсеребряный электрод представляет собой серебряную проволоку, покрытую слоем AgCl и помещенную в раствор KCl (3М) :
Хлорсеребряный электрод (ХСЭ) Ag, AgCl | Cl - представляет собой серебряный проводник, покрытый твердым AgCl, который погружается в насыщенный раствор KCl.
Серебро электрохимически взаимодействует со своим ионом:
|
Ag + + e - = Ag. |
|
Уравнение Нернста для этого процесса:
|
|
(7.2) |
Однако AgCl-активность - очень низкое содержание серебра. Но активность ионов Ag + связан с легко задаваемым в данной системе активности ионов Cl - произведение растворимость хлорид серебро ПР AgCl :
|
|
|
откуда
|
|
|
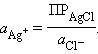
Подставляя это выражение в (7.2)
|
|
|
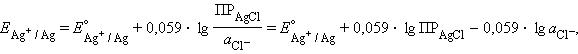
и обозначив
|
|
|
получим уравнение для хлорсеребряного электрода:
|
|
|
Электродный процесс может быть представлен уравнением
|
|
Потенциометрический метод анализа. Теоретические основы метода. Потенциал индикаторного электрода. Формальный потенциал. Классификация потенциометрических методов.
Потенциометрия – равновесный метод. *ΔG = 0; не нужен внешний источник энергии.
Потенциометрические методы основаны на измерении электродвижущих сил (ЭДС) – напряжения на электродах ячейки в отсутствии тока:
Е = Е1 – Е2, где
Е – электродвижущая сила; Е1 и Е2 – потенциалы электродов исследуемой цепи.
В этом случае один из электродов является неполяризуемым индикаторным электродом, а другой – электродом сравнения.
Для проведения потенциометрического анализа обычно собирают гальванический элемент. Гальванический элемент состоит из индикаторного электрода, потенциал которого зависит от концентрации (активности) определяемого иона, и электрода сравнения, потенциал которого должен оставаться постоянным независимо от протекания каких-либо реакций в анализируемом растворе. ЭДС исследуемого элемента выражается как разность между потенциалом электрода сравнения (Еср ) и потенциалом индикаторного электрода (Еинд ):
Е = Еср – Еинд + Ед , где Ед, – диффузионный потенциал, возникающий на границе между растворами двух разных электролитов или растворов разной концентрации одного электролита. На практике диффузионный потенциал стремятся элиминировать (свести к нулю) с помощью солевого мостика.
Потенциал индикаторного электрода связан уравнением Нернста с концентрацией или активностью определяемого иона.
𝐸=𝐸°+𝑅𝑇𝑛𝐹𝑙𝑛𝑎𝑀𝑒𝑛+
Ф ормальным
потенциалом принято
называть потенциал полуреакции при
условии, что концентрации окисленной
и восстановленной форм равны 1 М, а
концентрации посторонних электролитов
известны.
ормальным
потенциалом принято
называть потенциал полуреакции при
условии, что концентрации окисленной
и восстановленной форм равны 1 М, а
концентрации посторонних электролитов
известны.
Классификация потенциометрических методов:
Метод градуировочного графика. Готовят таким образом серию из 5–7 эталонных растворов с известным содержанием определяемого вещества. Концентрация определяемого вещества и ионная сила в эталонных растворах не должны сильно отличаться от концентрации и ионной силы ан-го раствора(так уменьшаются ошибки определения). Ионную силу всех растворов поддерживают постоянной введением индифферентного электролита (метод постоянной ионной силы). Эталонные растворы последовательно вносят в электрохимическую ячейку (стеклянный химический стакан, в который помещают электроды).
Измеряют ЭДС эталонных растворов, промывая дистиллированной водой электроды и стакан перед заполнением ячейки каждым эталонным раствором. По полученным данным строят градуировочный график в координатах Е(ЭДС) – lg c, где с – концентрация определяемого вещества в эталонном растворе. (Метод постоянной ионной силы позволяет перейти от активностей к концентрациям). Затем в электрохимическую ячейку вносят анализируемый раствор и измеряют ЭДС ячейки. По градуировочному графику находят lg c(Х), где c(Х) – концентрация определяемого вещества в анализируемом растворе.
По данным калибровки ИСЭ определяют следующие электрохимические характеристики.
1. Нернстовскую область электродной функции – интервал прямолинейной зависимости потенциала от активности (концентрации) потенциалопределяющих ионов.
2. Крутизну электродной функции – угловой коэф-нт наклона градуир-го графика (Е – раi, Е – рci).
3. Предел обнаружения потенциалопределяющего иона сmin. Для этого экстраполируют прямолинейные участки зависимости Е – рci; полученная точка пересечения соответствует на оси абсцисс величине сmiт .
4. Время отклика ИСЭ – время достижения стационарного потенциала.
5. Селективность электрода относительно определяемого иона в присутствии посторонних ионов.
Метод добавок стандарта. В электрохимическую ячейку вносят известный объем V(X) анализируемого раствора с концентрацией c(Х) и измеряют ЭДС ячейки. Затем в тот же раствор прибавляют точно измеренный небольшой объем стандартного раствора V(st) с известной (достаточно большой) концентрацией с(st) определяемого вещества и снова определяют ЭДС ячейки (необходимо, чтобы ∆Е ≥ 30 мВ).
Рассчитывают концентрацию
с(Х)
определяемого вещества в анализируемом
растворе по формул:
 ,
где ∆Е
– наблюдаемое изменение потенциала в
мВ после добавки стандарта; S
= 0.059/n
– крутизна электродной функции, В.
,
где ∆Е
– наблюдаемое изменение потенциала в
мВ после добавки стандарта; S
= 0.059/n
– крутизна электродной функции, В.
Классификация электродов. Функции индикаторных электродов и электродов сравнения.
Электродом сравнения называют электрод, потенциал которого постоянен и не зависит от концентрации ионов в растворе. Солевой мостик служит для предотвращения смешивания анализируемого раствора и раствора электрода сравнения.
Индикаторные электроды : должен удовлетворять ряду требований. Необходимо, чтобы его потенциал был воспроизводим и устанавливался достаточно быстро. При исследовании потенциала металлического электрода в растворе его соли индикаторный электрод должен быть обратим. Электрод должен обладать также определенной химической устойчивостью, чтобы не реагировать с другими компонентами анализируемого раствора. В потенциометрии в качестве индикаторных применяют металлические и мембранные (ионселективные) электроды.
Металлические электроды первого рода представляют собой металлическую пластинку или проволоку, погруженную в раствор хорошо растворимой соли этого металла.
Редокс-электроды служат для измерения окислительно-восстановительного потенциала системы. В качестве редокс-электродов используют платину, золото, иридий и графит. Потенциал этих электродов зависит от отношения активностей окисленной и восстановленной форм определяемого вещества (редокс-пары) и не зависит от материала электрода.
Электроды второго рода состоят из металла, покрытого слоем малорастворимого соединения этого металла и погруженного в раствор хорошо растворимого соединения с тем же анионом. К ним относятся хлорсеребряный, каломельный и некоторые другие
Ионселективные электроды (ИСЭ) основаны на использовании не электрохимической реакции с переносом электрона, а разности потенциалов, возникающей на границе раздела фаз, и равновесия обмена ионов между мембраной и раствором.
Функции индикаторных электродов сравнения: В потенциометрии применяют два основных класса индикаторных электродов: 1. Электроды, на межфазных границах которых протекают реакции с участием электронов, так называемые электронообменные (окислительно-восстановительные, электроды первого и второго рода). В оксилит- вост. электродов, роль электрода сводится к тому, что он является переносчиком электрона. Тот, кто окисляется он подходя к поверх. электрода отдает электроны электроду, а тот кто восстанавливается, он снимает эти электроны с поверх. электрода. Сам материал электрода не принимает участие в это процессе. 2. Электроды, на межфазных границах которых протекают ионообменные реакции. Такие электроды называют мембранными, или ионообменными, их называют также ионоселективными. Перенос заряда через межфазную границу осуществляется ионами. функция индикаторных электродов, измерить кол-во нашего исследуемого в-ва
Рассмотрим гальванический элемент Якоби-Даниэля. Он состоит из медной пластины, погруженной в раствор CuSО4, и цинковой пластины, погруженной в раствор ZnSО4. Для предотвращения прямого взаимодействия окислителя и восстановителя электроды отделены друг от друга пористой перегородкой. Схема гальванического элемента: Zn | ZnSO4| | CuSO4| Cu, Zn | Zn2+ | | Cu2+ | Cu. На поверхности цинковой пластины возникает двойной электрический слой и устанавливается равновесие:,Zn-2e « Zn2+. В результате протекания этого процесса возникает электродный потенциал цинка. На поверхности медной пластины также возникает двойной электрический слой и устанавливается равновесие: Сu2+ + 2е « Сu, поэтому возникает электродный потенциал меди. Потенциал цинкового электрода имеет более отрицательное значение, чем потенциал медного электрода, поэтому при замыкании внешней цепи, т. е. при соединении цинка с медью метал лическим проводником, электроны будут переходить от цинка к меди. Таким образом, при замыкании внешней цепи возникают самопроизвольные процессы растворения цинка на цинковом электроде и выделения меди на медном электроде. Данные процессы будут продолжаться до тех пор, пока не выровняются потенциалы электродов или не растворится весь цинк (или не высадится на медном электроде вся медь). Так же пока мы имеем дело с ионоселективными электродами, то в первую очередь мы снимаем электродную функцию ион-селективного электрода (это зависимость потенциала от логарифма активности иона, к которому чувствителен или обратим данный электрон). Так же не стоит забыть о методе постоянной ионной силы, который заключается в том, что снимаем зависимость не от логарифма активности, а от логарифма концентрации.
Ионселективные электроды. Различные виды ионселективных электродов. Уравнения для потенциалов ионселективных электродов.
Ионселективные электроды
Стеклянный электрод для измерения рН называются ионселективными. Многие ИСЭ аналогичны стеклянному. ИСЭ классифицируются по типу мембраны и подразделяются на твердые и жидкостные.
Твердые ИСЭ подразделяются на электроды с монокристаллическими и поликристаллическими мембранами.
В ИСЭ с монокристаллической мембраной ионочувствительный элемент изготавливается из малорастворимого кристаллического вещества с ионным характером проводимости. Перенос заряда в таком кристалле происходит за счет дефектов кристаллической решетки. Вакансии могут заниматься только ионом определенного размера и заряда, что обусловливает высокую селективность монокристаллических мембран. применяется фторидный электрод, в котором мембраной является LaF3, имеющий чисто фторидную проводимость.
ИСЭ с поликристаллическими мембранами обладают недостаточно устойчивым потенциалом и их селективность невелика. В таких электродах мембрана изготавливается путем смешивания активного вещества с инертной матрицей. ионселективный электрод с мембраной из сульфида серебра, пригодный для измерения активности и Ag+-, и S2--ионов. На основе сульфида серебра конструируются также различные галогенидные и металлочувствительные электроды.
Жидкостные ИСЭ имеют жидкую мембрану. В таких электродах раствор сравнения отделен от анализируемого тонким слоем органической жидкости, содержащей жидкий ионит, не смешивающийся с водой, но селективно реагирующий с определяемым ионом. Слой ионочувствительной органической жидкости получается пропиткой этой жидкостью пористой гидрофобной мембраны из пластика. Электроды такого типа существуют для Са2+, Na+, K+, NH4+.
Так же есть газочувствительные мембранные электроды для определения NH3, NO и др. газов. Используют пленочные электроды, в которых вместо жидкой мембраны используют тонкую пленку. У пленочных электродов такой же механизм действия, что и у мембранных, но они долговечнее и более удобны в работе.
ИСЭ можно охарактеризовать
количественно, используя уравнение
Никольского. Оно описывает зависимость
потенциала электрода от концентрации
посторонних ионов при помощи коэффициента
селективности:
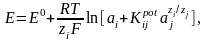
где аi – активность определяемого иона с зарядом zi; аj – активность определяемого иона с зарядом zj; Kpotij – потенциометрический коэффициент селективности.
при наличии m
мешающих ионов:
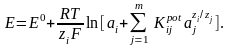
Устройство стеклянного электрода. Вывести уравнение, описывающее потенциал стеклянной мембраны. Достоинства и недостатки электрода.
Стеклянный электрод представляет собой тонкостенный стеклянный шарик 1, заполненный раствором HCl или каким-либo буферным раствором 2. Внутрь шарика помещают хлорсеребряный электрод 3. Это устройство обычно закрывают защитной трубкой 4.
При контакте с раствором приповерхностный слой стекла выступает в роли ионообменника, обменивая катионы, находящиеся в пустотах силикатного каркаса, на катионы водорода. Для того чтобы мембрана электрода приобрела такую способность, ее следует предварительно вымочить в кислом растворе.
Достоинства стеклянного электрода:
· простота работы;
· быстрое установление равновесия;
· широкая область определения рН;
· работает от 0º до 95ºС.
Недостатки стеклянного электрода:
· хрупкость;
· нельзя использовать обычную потенциометрическую установку вследствие большого сопротивления электрода;
· для усиления тока электродной пары со стеклянным электродом необходимо использовать струнные или зеркальные гальванометры или ламповые усилители;
· возможна погрешность в определении стандартного значения
Выввод ур-ия:
Ионы водорода внешней
стороны мембраны находятся в равновесии
с ионами водорода в исследуемом растворе
и на границе раздела возникает
потенциал Е1,
который определяется активностью ионов
водорода во внешнем растворе и на
поверхности геля:
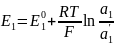
Потенциал Е2,
возникающий на границе внутреннего
раствора и геля, определяется активностью
ионов водорода во внутреннем растворе
(а2)
и на соответствующей поверхности геля
(а´2):
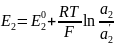
Граничный потенциал мембраны (Ем) стеклянного электрода определяется уравнением:
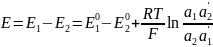 При постоянных
значениях а´1 и а´2 уравнение
принимает вид:
При постоянных
значениях а´1 и а´2 уравнение
принимает вид:

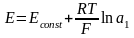 =
Econst
– 59,16pH
=
Econst
– 59,16pH
Селективность ионселективных электродов. Уравнение Никольского. Коэффициент селективности.
ИСЭ можно охарактеризовать количественно, используя уравнение Никольского. Оно описывает зависимость потенциала электрода от концентрации посторонних ионов при помощи коэффициента селективности. В случае одного постороннего иона уравнение Никольского выглядит так: где аi – активность определяемого иона с зарядом zi; аj – активность определяемого иона с зарядом zj; Kpotij – потенциометрический коэффициент селективности.
В общем случае, при наличии m мешающих ионов:
Коэффициент селективности показывает, при каком соотношении концентраций определяемого и постороннего ионов последний начинает оказывать мешающее влияние. Значения коэффициентов селективности изменяются от весьма малых величин, близких к нулю, до единицы и более. Чем меньше величина коэффициента селективности, тем выше селективность электрода.
Наиболее предпочтительным способом определения коэффициента селективности является способ, основанный на исследовании растворов, содержащих смеси определяемого и постороннего иона. Для этого получают серии градуировочных зависимостей потенциала электрода от концентрации определяемого иона, построенных в присутствии различных концентраций постороннего иона (рис.).
При больших избытках посторонних ионов потенциал электрода определяется вторым слагаемым в уравнении Никольского. Он остается постоянным и не зависит от концентрации определяемого иона. Из абсциссы точки пересечения двух уч ai =Kijpot∙ajz(i) / z(j), зная постоянную величину aj, рассчитать коэффициент селективности. Описанный метод позволяет рассчитать коэффициент селективности даже по одной градуировочной зависимости, но для большей достоверности надо использовать несколько.
Прямая потенциометрия. Основные приемы, используемые в прямой потенциометрии. Область применения метода.
Методы прямой потенциометрии (ионометрии) основаны на непосредственном применении уравнения Нернста для нахождения активности или концентрации участника электродной реакции по экспериментально измеренной ЭДС цепи или потенциалу электрода.
Классификация методов:
Метод градуировочного графика. Готовят таким образом серию из 5–7 эталонных растворов с известным содержанием определяемого вещества. Концентрация определяемого вещества и ионная сила в эталонных растворах не должны сильно отличаться от концентрации и ионной силы ан-го раствора(так уменьшаются ошибки определения). Ионную силу всех растворов поддерживают постоянной введением индифферентного электролита (метод постоянной ионной силы). Эталонные растворы последовательно вносят в электрохимическую ячейку (стеклянный химический стакан, в который помещают электроды).
Измеряют ЭДС эталонных растворов, промывая дистиллированной водой электроды и стакан перед заполнением ячейки каждым эталонным раствором. По полученным данным строят градуировочный график в координатах Е(ЭДС) – lg c, где с – концентрация определяемого вещества в эталонном растворе. (Метод постоянной ионной силы позволяет перейти от активностей к концентрациям). Затем в электрохимическую ячейку вносят анализируемый раствор и измеряют ЭДС ячейки. По градуировочному графику находят lg c(Х), где c(Х) – концентрация определяемого вещества в анализируемом растворе.
По данным калибровки ИСЭ определяют следующие электрохимические характеристики.
1. Нернстовскую область электродной функции – интервал прямолинейной зависимости потенциала от активности (концентрации) потенциалопределяющих ионов.
2. Крутизну электродной функции – угловой коэф-нт наклона градуир-го графика (Е – раi, Е – рci).
3. Предел обнаружения потенциалопределяющего иона сmin. Для этого экстраполируют прямолинейные участки зависимости Е – рci; полученная точка пересечения соответствует на оси абсцисс величине сmiт .
4. Время отклика ИСЭ – время достижения стационарного потенциала.
5. Селективность электрода относительно определяемого иона в присутствии посторонних ионов.
Метод добавок стандарта. В электрохимическую ячейку вносят известный объем V(X) анализируемого раствора с концентрацией c(Х) и измеряют ЭДС ячейки. Затем в тот же раствор прибавляют точно измеренный небольшой объем стандартного раствора V(st) с известной (достаточно большой) концентрацией с(st) определяемого вещества и снова определяют ЭДС ячейки (∆Е ≥ 30 мВ).
Рассчитывают концентрацию с(Х) определяемого вещества в анализируемом растворе по формуле
,
где ∆Е – наблюдаемое изменение потенциала в мВ после добавки стандарта; S = 0.059/n – крутизна электродной функции, В.
Этот метод позволяет проводить быстрее и точнее определение концентрации или активности ионов и обладающих рядом других достоинств.
Потенциометрическое титрование. Различные способы определения точки эквивалентности.
ПТТ – способ определения объема титранта, затраченного на титрование определяемого вещества в анализируемом растворе, путем измерения ЭДС) с помощью гальванической цепи, составленной из индикаторного электрода и электрода сравнения. При ПТТ анализируемый раствор, находящийся в электрохимической ячейке, титруют подходящим титрантом. Конечную точку титрования (КТТ) находят по скачку потенциала индикаторного электрода, отвечающему моменту завершения реакции. При ПТТ не требуется использование индикаторов, изменяющих окраску вблизи точки эквивалентности (ТЭ).
Титрант прибавляют равными порциями, каждый раз измеряя разность потенциалов. В конце титрования (вблизи ТЭ) титрант прибавляют по каплям, также измеряя разность потенциалов.
Кривые потенциометрического титрования. Кривая потенциометрического титрования – графическое изображение изменения ЭДС электрохимической ячейки в зависимости от объема прибавленного титрнта.
Кривые ПТТ
Е
 – V – интегральные кривые титрования:
кривая титрования сильной кислоты
сильным основанием. В точке эквивалентности
происходит резкий скачок ЭДС, вызванный
резким изменением потенциала индикаторного
электрода. По этому скачку можно
определить точку эквивалентности и
рассчитать содержание кислоты.
– V – интегральные кривые титрования:
кривая титрования сильной кислоты
сильным основанием. В точке эквивалентности
происходит резкий скачок ЭДС, вызванный
резким изменением потенциала индикаторного
электрода. По этому скачку можно
определить точку эквивалентности и
рассчитать содержание кислоты.
∆
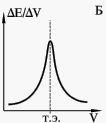 Е/∆V
– V – дифференциальные
кривые
титрования по первой производной:
Для
нахождения точки эквивалентности. На
точку эквивалентности указывает
максимум полученной кривой, а отсчет
по оси абсцисс, соответствующий этому
максимуму, дает объем титранта,
израсходованного на титрование до
точки эквивалентности. Определение
точки эквивалентности по дифференциальной
кривой значительно точнее, чем по
простой зависимости E - V.
Е/∆V
– V – дифференциальные
кривые
титрования по первой производной:
Для
нахождения точки эквивалентности. На
точку эквивалентности указывает
максимум полученной кривой, а отсчет
по оси абсцисс, соответствующий этому
максимуму, дает объем титранта,
израсходованного на титрование до
точки эквивалентности. Определение
точки эквивалентности по дифференциальной
кривой значительно точнее, чем по
простой зависимости E - V.
∆
 2
Е/∆V 2
– V – дифференциальные кривые титрования
по второй производной:
Поскольку
производная функции, имеющей максимум,
в точке максимума равна нулю, вторая
производная потенциала по объему
(d2E/dV2) в точке эквивалентности будет
равна нулю. Это свойство также используется
для нахождения точки эквивалентности
2
Е/∆V 2
– V – дифференциальные кривые титрования
по второй производной:
Поскольку
производная функции, имеющей максимум,
в точке максимума равна нулю, вторая
производная потенциала по объему
(d2E/dV2) в точке эквивалентности будет
равна нулю. Это свойство также используется
для нахождения точки эквивалентности
∆V/∆Е – V – по методу Грана: Перед точкой эквивалентности и после нее кривая Грана линейна, а сама точка эквивалентности находится как точка пересечения этих прямых. Достоинства и удобства метода Грана особенно заметны при анализе разбавленных растворов, позволяя определить точку эквивалентности с достаточной точностью вследствие линейности графика.
где Е – ЭДС потенциометричской ячейки, V – объем прибавленного титранта, ∆Е – изменение потенциала, соответствующее прибавлению ∆V титранта; ∆Е/∆V ≈ dE/dV;
∆ 2 Е/∆V 2 = ∆(∆Е)/(∆V) ≈ d 2 E/dV 2 .
Теоретические основы кондуктометрического метода анализа.

· прямая кондуктометрия - метод, позволяющий непосредственно определять концентрацию электролита путем измерения электропроводности раствора с известным качественным составом;
· кондуктометрическое титрование - метод анализа, основанный на определении содержания вещества по излому кривой титрования. Кривую строят по измерениям удельной электропроводности анализируемого раствора, меняющейся в результате химических реакций в процессе титрования

Эквивалентной электропроводностью называют проводимость раствора, содержащего 1 моль эквивалента вещества и находящегося между двумя параллельными электродами, расстояние между которыми 1 см; единица измерения См∙см2/моль-экв.
Удельная и эквивалентная электропроводности связаны между собой: λ = 1000æ/с
где λ – эквивалентная электропроводность; с – молярная концентрация эквивалента.
Молярная и эквивалентная электропроводности определяются величинами подвижностей ионов. Подвижность является характеристикой движения ионов в электрическом поле, не зависящей от напряженности поля: u = v/E,
где u – подвижность иона; v – скорость движения иона в электрическом поле; E – напряженность электрического поля.
Связь между эквивалентной электропроводностью и подвижностями ионов (u+ – подвижность катионов, u- – подвижность анионов) следующая: λ = F(u+ + u-) = λ+ + λ
Это уравнение отражает закон независимого движения ионов – закон Кольрауша: в растворе электролита катионы и анионы проводят электрический ток независимо друг от друга.
Электропроводность растворов (удельная, эквивалентная и молярная), ее зависимость от природы электролита и растворителя, концентрации сильного и слабого электролита и от температуры.
Эквивалентной электропроводностью называют проводимость раствора, содержащего 1 моль эквивалента вещества и находящегося между двумя параллельными электродами, расстояние между которыми 1 см; единица измерения См∙см2/моль-экв.
Удельная и эквивалентная электропроводности связаны между собой: λ = 1000æ/с
где λ – эквивалентная электропроводность; с – молярная концентрация эквивалента.
Молярная и эквивалентная электропроводности определяются величинами подвижностей ионов. Подвижность является характеристикой движения ионов в электрическом поле, не зависящей от напряженности поля: u = v/E,
где u – подвижность иона; v – скорость движения иона в электрическом поле; E – напряженность электрического поля.
Связь между эквивалентной электропроводностью и подвижностями ионов (u+ – подвижность катионов, u- – подвижность анионов) следующая: λ = F(u+ + u-) = λ+ + λ
Это уравнение отражает закон независимого движения ионов – закон Кольрауша: в растворе электролита катионы и анионы проводят электрический ток независимо друг от друга.
Электропроводность определяется также степенью диссоциации электролита. Для сильных и слабых электролитов зависимость электропроводности от концентрации различна.
У полностью диссоциированных сильных электролитов в области разбавленных растворов (0.001 М и меньше) концентрационная зависимость электропроводности выражается уравнением : λ = λ0 – а 1/ 2
где λ0 – предельная эквивалентная электропроводность сильного электролита при бесконечном разбавлении; а – константа.
Числовые значения подвижностей ионов в водном растворе при комнатной температуре находятся в пределах 30–70 См∙см2/моль-экв. Лишь у ионов Н+ и ОН- они существенно выше, что связано с особым эстафетным механизмом перемещения этих ионов в электрическом поле.
Для слабых электролитов концентрационная зависимость электропроводности имеет более сложный характер. Это объясняется тем, что на λ в этом случае влияет еще и увеличение степени диссоциации электролита с разбавлением раствора, вызывающее более быстрое, чем у сильных электролитов, увеличение электропроводности.
Электроды, используемые в кондуктометрии, изготавливают из платины, серебра или нержавеющей стали. Токопроводящим является не только тот объем раствора, который заключен между электродами.
Электропроводность сильных электролитов. Эффекты электрофоретического и релаксационного торможения по теории Дебая-Онзагера.
У полностью диссоциированных (сильных) электролитов в области разбавленных растворов (0.001 М и меньше) концентрационная зависимость проводимости выражается уравнением:

С уменьшением концентрации электролита эквивалентная электрическая проводимость возрастает и в области бесконечного разбавления стремится к предельному значению экв. электрической проводимости λ0, характеризующему электрическую проводимость гипотетически бесконечно разбавленного раствора.

Оба эффекта связаны с существованием вокруг иона ионной атмосферы из противоположно заряженных ионов.

Электропроводность слабых электролтов.
Слабые электролиты в растворе диссоциируют только частично, к ним относятся почти все органические кислоты и основания (СН3СООН, СН3СООNa), а также соли сильных кислот и слабых оснований и наоборот, сильных оснований и слабых кислот.
Электропроводность определяется также степенью диссоциации электролита. Для сильных и слабых электролитов зависимость электропроводности от концентрации различна.
У полностью диссоциированных сильных электролитов в области разбавленных растворов (0.001 М и меньше) концентрационная зависимость электропроводности выражается уравнением : λ = λ0 – а 1/ 2
где λ0 – предельная эквивалентная электропроводность сильного электролита при бесконечном разбавлении; а – константа.
При увеличении концентрации и сильных, и слабых электролитов увеличивается количество носителей зарядов (ионов). В разбавленных растворах это приводит к практически линейному росту удельной электропроводности с концентрацией. Однако, в растворах слабых электролитов, где степень диссоциации a << 1, число ионов и æ увеличивается медленнее. У слабых электролитов форма концентрационной зависимости эквивалентной электропроводности определяется изменением степени диссоциации электролитов.
Инструментальная база кондуктометрических методов анализа. Схема и принцип работы моста переменного тока.
 Это
мостик Уитстона, питаемый переменным
током от генератора. Переменного тока
не должно быть, тк произойдет эл-з.
Применение моста переменного тока
приводит к появлению реактивного
сопротивления Rx.
Поэтому нельзя свести к нулю силу тока
в диагонали моста (гв). Положение
передвижного контакта (в) подбирается
так, чтобы нуль-инструмент 1 не показывал
ток . тогда сопр-ие Rx
:
Это
мостик Уитстона, питаемый переменным
током от генератора. Переменного тока
не должно быть, тк произойдет эл-з.
Применение моста переменного тока
приводит к появлению реактивного
сопротивления Rx.
Поэтому нельзя свести к нулю силу тока
в диагонали моста (гв). Положение
передвижного контакта (в) подбирается
так, чтобы нуль-инструмент 1 не показывал
ток . тогда сопр-ие Rx
:
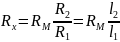 , где l1
и
l2
– длины плеч
реохорда при компенсации, пропорц-ые
сопр-ям R1
(ав) и R2
(вб)
, где l1
и
l2
– длины плеч
реохорда при компенсации, пропорц-ые
сопр-ям R1
(ав) и R2
(вб)
Конструкции измерительных электрохимических ячеек весьма разнообразны. В прямой кондуктометрии обычно используют ячейки с жестко закрепленными в них электродами. В методах кондуктометрического титрования – ячейки с погруженными электродами, позволяющие проводить титрование в любых сосудах, в которых можно разместить электроды. Электроды, используемые в кондуктометрии, изготавливают из платины, серебра или нержавеющей стали.
Экспериментально измеряемая величина сопротивления раствора зависит не только от размера электродов и расстояния между ними, но и от их формы и взаимного расположения, объема раствора и других факторов. Действительная электрическая проводимость раствора не зависит от этих факторов, а определяется лишь концентрацией раствора, природой электролита и температурой. Истинная электрическая проводимость пропорциональна экспериментально измеренной величине проводимости: æ = kæI
где k – константа ячейки.
Константу ячейки находят экспериментально по электрической проводимости стандартных растворов с хорошо известным значением электропроводности в широкой области температур и концентраций. Обычно используют растворы хлорида калия.
Прямая и косвенная кондуктометрия. Контактная и неконтактная кондуктометрия. Области применения.
Методы прямой кондуктометрии основаны на том, что в области разбавленных и умеренно концентрированных растворов электрическая проводимость растет с увеличением концентрации электролита. В практической работе обычно используют заранее построенную градуировочную кривую зависимости электропроводности от концентрации тех или иных электролитов. В связи с относительно близкими значениями подвижностей ионов кондуктометрические измерения дают информацию, главным образом, лишь об общей концентрации ионов в растворе. Малая селективность кондуктометрических методов является одним из его существенных недостатков.
В методах кондуктометрического титрования (косвенная кондуктометрия) измеряют электрическую проводимость раствора после добавления небольших определенных порций титранта и находят точку эквивалентности графическим методом с помощью кривой в координатах æ –V(титранта). Практически в этом методе могут быть использованы такие химические реакции, в ходе которых достаточно заметно изменяется электропроводность раствора или происходит резкое изменение электропроводности после точки эквивалентности.
Это такие реакции, как:
1.Реакции кислотно-основного взаимодействия
2. Реакции осаждения
3. Реакции комплексообразования
Различают контактную и бесконтактную кондуктометрию в зависимости от наличия или отсутствия контакта между электролитом и входными цепями измерит. прибора. наиб. распространены контактный низкочастотный и бесконтактный высокочастотный методы.
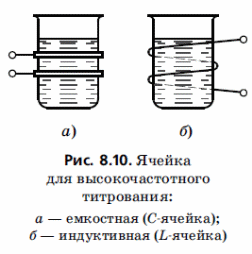 В
контактной низкочастотной кондуктометрии
используют контактные электрохимические
ячейки, в которые между двумя инертными
электродами набирается какое-то
количество электролита. Электроды имеют
удельную площадь поверхности, они
находятся на определённом расстоянии
друг от друга. Для мостов переменного
тока это имеет большое значение.
В
контактной низкочастотной кондуктометрии
используют контактные электрохимические
ячейки, в которые между двумя инертными
электродами набирается какое-то
количество электролита. Электроды имеют
удельную площадь поверхности, они
находятся на определённом расстоянии
друг от друга. Для мостов переменного
тока это имеет большое значение.
Ячейки с анализируемым раствором при высокочастотном титровании помещается или между пластинками конденсатора, или внутри индукционной катушки
В первом случае ячейку называют конденсаторной или ёмкостной, или С-ячейкой, а во втором – индуктивной или L-ячейкой. В ячейках высокочастотного титрования электроды не соприкасаются с исследуемым раствором, что является одним из существенных достоинств метода.

Кондуктометрическое определение физико-химических свойств и характеристик веществ с помощью прямой кондуктометрии. Определение константы диссоциации слабой одноосновной кислоты, если табличные данные о подвижности ионов отсутствуют.


Кондуктометрическое титрование. Виды реакций, лежащих в основе кондуктометрического титрования. Вид кривых титрования.
В методах кондуктометрического титрования измеряют электрическую проводимость раствора после добавления небольших определенных порций титранта и находят точку эквивалентности графическим методом с помощью кривой в координатах æ –V(титранта). Практически используют такие химические реакции, в ходе которых достаточно заметно изменяется электропроводность раствора или происходит резкое изменение электропроводности после точки эквивалентности.
Реакции кислотно-основного взаимодействия
П ри
титровании сильной
кислоты сильным основанием
до ТЭ наблюдается резкое уменьшение
электрической проводимости, связанное
с уменьшением в растворе концентрации
иона Н+.
После ТЭ начинается резкий подъем
электропроводности, так как будет расти
концентрация ионов ОН-
ри
титровании сильной
кислоты сильным основанием
до ТЭ наблюдается резкое уменьшение
электрической проводимости, связанное
с уменьшением в растворе концентрации
иона Н+.
После ТЭ начинается резкий подъем
электропроводности, так как будет расти
концентрация ионов ОН-
Для построения используются области недотитрованных и перетитрованных растворов. При титровании очень разбавленных растворов, обе ветви кривой титрования сглаживаются, и ТЭ может быть определена только с помощью экстраполяции прямолинейных участков кривой.
К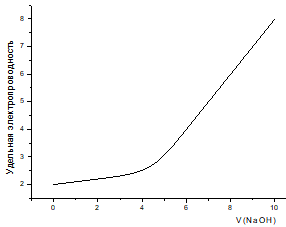 ривая
титрования слабой
кислоты сильным основанием.
Удельная электрическая проводимость
раствора до ТЭ несколько возрастает,
т. к. при титровании происходит увеличение
концентрации соли. Концентрация ионов
Н+
в растворе слабой кислоты и ее соли,
получившейся при титровании, невелика
и уменьшение [Н+]
не вызывает резкого падения
электропроводности.
ривая
титрования слабой
кислоты сильным основанием.
Удельная электрическая проводимость
раствора до ТЭ несколько возрастает,
т. к. при титровании происходит увеличение
концентрации соли. Концентрация ионов
Н+
в растворе слабой кислоты и ее соли,
получившейся при титровании, невелика
и уменьшение [Н+]
не вызывает резкого падения
электропроводности.
к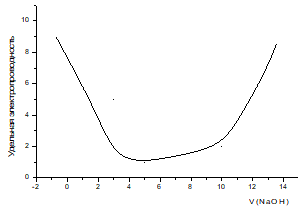 ривая
титрования смеси
сильной и слабой кислот.
Кривая имеет два излома, соответствующие
двум точкам эквивалентности: первая
показывает объем щелочи, пошедшей на
титрование сильной кислоты, а вторая
дает объем щелочи, израсходованный на
титрование обеих кислот. С увеличением
константы диссоциации слабой кислоты
первый излом будет становиться менее
резким, а второй – более четким, и,
наоборот, чем слабее кислота, тем резче
будет первый излом, и более закругленным
второй.
ривая
титрования смеси
сильной и слабой кислот.
Кривая имеет два излома, соответствующие
двум точкам эквивалентности: первая
показывает объем щелочи, пошедшей на
титрование сильной кислоты, а вторая
дает объем щелочи, израсходованный на
титрование обеих кислот. С увеличением
константы диссоциации слабой кислоты
первый излом будет становиться менее
резким, а второй – более четким, и,
наоборот, чем слабее кислота, тем резче
будет первый излом, и более закругленным
второй.
Реакции осаждения
Вид кривой осаждения зависит от концентрации и подвижности ионов и растворимости образующегося соединения. Чем меньше ПР продукта реакции, тем резче выражен излом кривой титрования в ТЭ. У более растворимых соединений точка эквивалентности устанавливается с трудом, т. к. кривая титрования плавно закругляется. Излом на кривой титрования становится менее четким также с уменьшением концентрации раствора. Введение в анализируемый водный раствор органического растворителя понижает растворимость, поэтому излом на кривой титрования становится более резким.
Влияние подвижности ионов проявляется в наклоне кривой титрования до точки эквивалентности. После точки эквивалентности проводимость во всех случаях будет расти, т. к. увеличивается концентрация ионов в растворе.
Реакции комплексообразования
Для кондуктометрического
титрования катионов в качестве титрантов
могут быть использованы растворы
различных кислот и оксикислот (щавелевой,
винной, лимонной и др.), комплексонов и
других лигандов. Н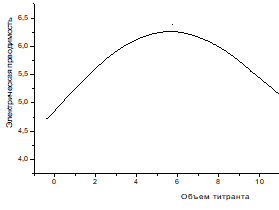 аибольшее
практическое значение имеет
кондуктометрическое титрование катионов
металлов двузамещенной солью
этилендиаминтетрауксусной кислоты
(ЭДТА).
аибольшее
практическое значение имеет
кондуктометрическое титрование катионов
металлов двузамещенной солью
этилендиаминтетрауксусной кислоты
(ЭДТА).
выделяются ионы Н+ и растет электрическая проводимость раствора. После точки эквивалентности электропроводность падает, т. к. выделившиеся ионы Н+ связываются анионом H2Y2-
Н+ + H2Y2- = H3Y- .
Кривая титрования катиона в буферном растворе имеет иной вид кривой титрования слабой кислоты сильным основанием. До ТЭ электрическая проводимость раствора несколько увеличивается, что связано с увеличением концентрации ионов натрия, вводимых с титрантом, а после ТЭ резко возрастает, т. к. увеличивается концентрация титранта.
Вольтамперометрия. Теоретические основы метода. Достоинства и недостатки метода.
Осн-на на изучении поляризац-х или вольтамперных кривых (зависимости силы тока от напряжения), которые получ-ся, если при эл-зе р-ра ан-го в-ва постепенно повышать напряжение и фиксировать при этом силу тока. В эл-зе исп-ют легко поляр-гося эле-д с малой поверх-тью, на котором происходит эл-восст-ние или эл-окисл в-ва, а пот-л неполяриз-ся эл-да сравнения остается постоянным.
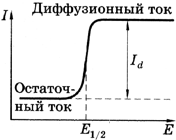 В
начале процесса при небольшом пот-ле
катода сила тока медленно увелич-ся с
возр-ем пот-ла (Это остаточный
ток). По достижении
пот-ла восст-ния на катоде начинается
разряд ионов и сила тока резко возр-т,
стремясь к предельной величине диффуз-го
тока. Конц-ия разряж-хся ионов в
прикатодном слое близка нулю. Конц-ия
разряж-ся иона в глубине р-ра постоянна,
т. к. эл-з идет при очень небольшой силе
тока. Поэтому разность конц-ий, опр-щая
ск-ть диффузии при данной темп-ре, будет
постоянна, что и приводит к постоянной
ск-ти поступления ионов к катоду. Это
состояние равновесия будет хар-ся
постоянной силой тока, не изм-ся при
увелич напряжения. Этот постоянный ток,
контр-мый диффузией, и наз-т диффуз-ым
током.
В
начале процесса при небольшом пот-ле
катода сила тока медленно увелич-ся с
возр-ем пот-ла (Это остаточный
ток). По достижении
пот-ла восст-ния на катоде начинается
разряд ионов и сила тока резко возр-т,
стремясь к предельной величине диффуз-го
тока. Конц-ия разряж-хся ионов в
прикатодном слое близка нулю. Конц-ия
разряж-ся иона в глубине р-ра постоянна,
т. к. эл-з идет при очень небольшой силе
тока. Поэтому разность конц-ий, опр-щая
ск-ть диффузии при данной темп-ре, будет
постоянна, что и приводит к постоянной
ск-ти поступления ионов к катоду. Это
состояние равновесия будет хар-ся
постоянной силой тока, не изм-ся при
увелич напряжения. Этот постоянный ток,
контр-мый диффузией, и наз-т диффуз-ым
током.
Вольтамперометрический метод достаточно универсален и применим к многочисленному кругу объектов. Основными достоинствами метода являются быстрота анализа, возможность определения нескольких веществ в смеси без предварительного разделения, достаточно высокая точность и применимость к анализу небольших содержаний определяемого элемента. При сочетании вольтамперометрии с методами экстракции, хроматографии и т.д. предел обнаружения снижается ещё больше.
Недостатки.
Для полярографии: токсичность капельного ртутного электрода; невозможность работать в анодной области (положительные потенциалы рабочего электрода, а так как для ртути величина стандартного потенциала равна 0,3В, то при дальнейшем увеличении потенциала ртуть начинает растворятся и анализ становится невозможным).
Теория классической полярографии
М-ды прямой полярографии осн-ны на непоср-ном применении ур-ий полярографической волны и Ильковича. Пот-л полуволны не зависит от конц-ии и явл-ся кач-ной хар-кой в-ва. Е1/2 опр-т графич-м м-дом. Уравнение E = E1/2 +(RT/nF)ln((Id –I)/I) показывает, что lg((Id –I)/I) явл-ся линейной ф-цией Е, и графическое отображение ф-ции Е = f (lg((Id –I)/I является прямой, которая пересекает ось абсцисс в точке Е = Е1/2.
Для идентификации неизв-го в-ва можно этим м-дом опр-ть пот-л полуволны, пользуясь табл, уст-ть наиболее вероятный эл-нт.
Для решения задач кол-го ан-за чаще всего прим-т м-д градуир-го графика. Коэф-нт пропорц-ти между конц-ей в-ва и силой диффуз-го тока в ур-нии Ильковича (Id = 605zD1/2m2/3t1/6c) обычно уст-т с помощью станд р-ров. При постоянных условиях полярографирования D, m и t постоянны, поэтому: Id = kc
Градуир-ый график строят по данным полярографирования нескольких станд-х р-ров. На оси ординат откл-ся пропорц-ая силе диффуз-го тока высота полярографич волны, а по оси абсцисс – конц-ия ан-го в-ва. В соотв-ии с ур-ем (Id = kc) градуировочный график должен представлять собой прямую линию, проходящую через начало координат. М-д дает точные рез-ты при условии строгой идент-ти условий полярографирования станд-х р-ров и неизвестной пробы.
 Схема
полярографической установки
Схема
полярографической установки
Ан-ый р-р 2 нах-ся в электролизере 3, на дне которого имеется слой ртути 1, явл-ся анодом. Катодом служит ртутный капающий эл-д 4, соед-ный с резервуаром ртути 5. Внешнее напряжение, подаваемое на эл-ды, можно плавно менять с помощью реохорда 7 и измерять при этом гальванометром 6 силу тока, проходящего через р-р.
Напряжение, которое подается на эл-ды, будет практ-ки целиком опр-ть пот-л ртутного кап-го катода.
В вольтамперометрии широко прим-т также твердые микроэл-ды, изготавл-ые из благородных ме или графита. Основным их достоинством является возм-ть работы в более положительной области пот-лов, чем с ртутным эл-дом.
Большое преим-во твердых микроэл-дов закл-ся в их нетоксичности. Неоспоримым преим-вом ртутного капающего эл-да явл-ся постоянное возобновление поверх-ти.
Так же прим-ют вращ-ся и вибрир-ие платиновые микроэл-ды, на которых устойчивая сила тока устанавл-ся быстро. При работе таких эл-дов р-р непрерывно перемеш-ся, благодаря чему к поверх-ти эл-да ионы доставл-ся не только за счет диффузии, но и за счет мех-го перемеш-ия. Это знач-но увелич-т предельный ток по сравнению с диффуз-м.
Как взаимосвязаны потенциал полуволны и предельный диффузионный ток.
З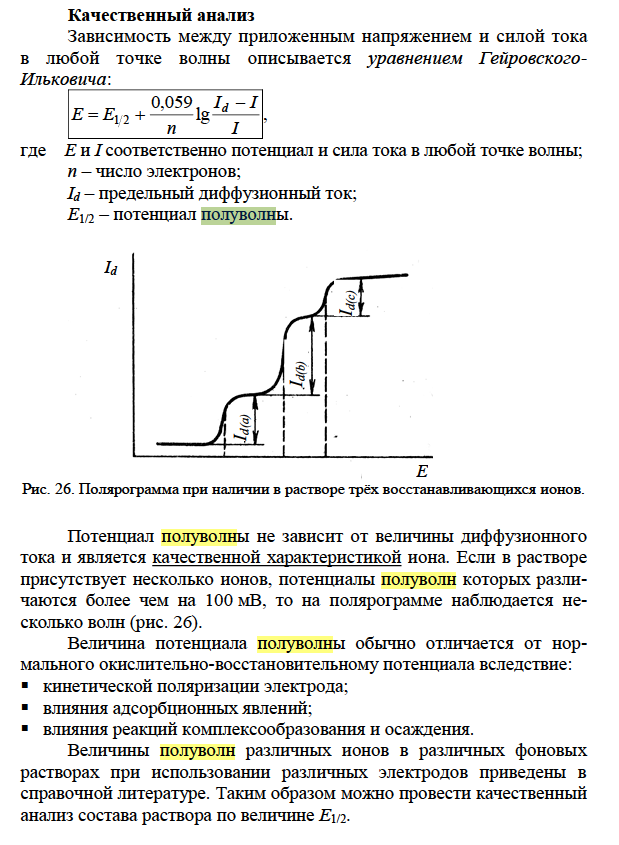 авис-ть
силы тока I
от прилож-го напряжения Е
при обратимом эл-дном процессе отражается
ур-ем полярографической волны:
E
= E1/2
+(RT/nF)ln((Id
–I)/I),
авис-ть
силы тока I
от прилож-го напряжения Е
при обратимом эл-дном процессе отражается
ур-ем полярографической волны:
E
= E1/2
+(RT/nF)ln((Id
–I)/I),
где Е1/2 – пот-л полуволны; Id – диффузионный ток.
В начале процесса при небольшом Е, I растет медленно (это остаточный ток), после достижения Е восстановления на катоде начинается разряд и I резко возрастает. Стремясь к предельной величине Id. Если I =1/2Id , то уравнение переходит в Е = Е1/2 – это соотношение показывает независимость потенциала полуволны от I и, следовательно, от концентрации восстанавливающегося иона. Потенциал полуволны яв-ся качественной характеристикой при данном фоновом электролите. Определение Е1/2 – основа качественного полярографического анализа. Е1/2 зависит от:
рН раствора;
2)природы;
3)конц. фонового электролита На вольтамперограмме можно наблюдать несколько волн.
На чем основан качественный полярографический анализ.
Основу качественного полярографического анализа составляет определение потенциала полуволны 𝐸12⁄°. Потенциал полуволны является качественной характеристикой иона.
Потенциал полуволны зависит от следующих факторов:
природы электрода;
природы разряжающихся ионов;
природы фона;
наличие в растворе комплексообразователей;
температуры.
Особое значение имеет наличие в растворе веществ, способных к комплексообразованию с определяемым ионом.
Сдвиг потенциала полуволны при введении в раствор лиганда значительно расширяет возможности полярографического анализа, позволяя создавать условия для определения нескольких компонентов в одном растворе без предварительного разделения. Если в растворе находится несколько веществ, потенциалы полуволны которых различаются на 100 мВ и больше, то на полярограмме будет не одна волна, а несколько – по числу восстанавливающихся ионов (рисунок) а возможно и больше, так как при ступенчатом восстановлении один ион может давать две волны.
Уравнения полярографической волны. Уравнение Ильковича.
Зависимость силы тока I от приложенного напряжения Е при обратимом электродном процессе отражается уравнением полярографической волны: 𝐸=𝐸12⁄°+𝑅𝑇𝑛𝐹ln𝐼𝑑−𝐼𝐼, где
𝐸12⁄° - потенциал полуволны, 𝐼𝑑 – диффузионный ток.
Типичная зависимость силы тока от приложенного напряжения дана на рисунке:
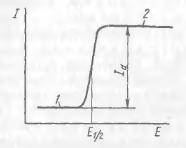
Рис.1 Полярограмма. 1-остаточный ток;
2-диффузионный ток
Из рисунка видно, что в начале процесса при небольшом потенциале катода сила тока медленно увеличивается с возрастанием потенциала – это так называемый остаточный ток. По достижении потенциала восстановления на катоде начинается разряд ионов, и сила тока резко возрастает, стремясь к предельной величине диффузионного тока.
Связь диффузионного тока 𝐼𝑑 с концентрацией иона cM и другими величинами передается уравнением Ильковича: 𝐼 𝑑= 605𝑧𝐷12⁄𝑚23⁄𝑡16⁄𝑐𝑀 , где
z – заряд иона; D – коэффициент диффузии; m – масса ртути, вытекающей из капилляра в 1 с, мг;
t – время образования капли (период капания).

Количественный полярографический анализ.
Количественный полярографический анализ основан на зависимости предельного диффузионного тока от концентрации ионов в растворе, которая математически даётся уравнением Ильковича: Id = 605zD1/2m2/3t1/6c,
где z – заряд иона, D – коэф-нт диффузии, m – масса ртути, вытек-ей из капилляра за одну секунду, мг, t – время образования капли (период капания), с.
В основе количественного полярографического анализа лежит уравнение Ильковича
Расчетный метод в основе лежит уравнению Ильковича м-д закл в измерение величины предельного диффузионного тока секундомером находят t, опр-т m, a D берут из табл и прямой расчет концентрации определяемого компонента но уравнению Ильковича: Id = 605zD1/2m2/3t1/6c,
М-д имеет ограничение применения, т.к. D известны .?. небольшого кол-ва эл-ов
метод стандартов. При
одинак условиях снимают
полярограммы 2-3 станд
р-ров и анализ р-ра. Графически нах-т все
Н:
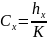
где Сст - концентрация раствора стандартного образца, мг/мл; hх - высота волны анализируемого раствора, мм; Hст - высота волны раствора стандартного образца, мм.
М-д градуир гр-ка. В основе ур-ие Id=kCpMeut. Готовят 5-6 станд р-ров. При одинак усл-х снимают полярограмму и находят h всех р-ров, строят график h=F(CpMeut) – это прямая линия. При тех же условиях снимают полярограмму ан-го р-ра ,измеряют hх и по графику нах-т Сх
М-д хорош при усл-х (одинак для всех р-ров):
Условия работы .?.
Темп-ра
Фон
метод добавок. Для опр-ия конц. дост-но снять 2 полярограммы :1- анализ р-р; 2- анализ р-р +доб-ки. Берем фиксир-ем V и снимаем завис-ть затем доб-ем доб-ку:
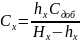
Различные приемы (методы), используемые в количественном полярографическом анализе.
В основе количественного полярографического анализа лежит уравнение Ильковича
Расчетный метод в основе лежит уравнению Ильковича м-д закл в измерение величины предельного диффузионного тока секундомером находят t, опр-т m, a D берут из табл и прямой расчет концентрации определяемого компонента но уравнению Ильковича: Id = 605zD1/2m2/3t1/6c,
М-д имеет ограничение применения, т.к. D известны .?. небольшого кол-ва эл-ов
метод стандартов. При одинак условиях снимают полярограммы 2-3 станд р-ров и анализ р-ра. Графически нах-т все Н:
где Сст - концентрация раствора стандартного образца, мг/мл; hх - высота волны анализируемого раствора, мм; Hст - высота волны раствора стандартного образца, мм.
М-д градуир гр-ка. В основе ур-ие Id=kCpMeut. Готовят 5-6 станд р-ров. При одинак усл-х снимают полярограмму и находят h всех р-ров, строят график h=F(CpMeut) – это прямая линия. При тех же условиях снимают полярограмму ан-го р-ра ,измеряют hх и по графику нах-т Сх
М-д хорош при усл-х (одинак для всех р-ров):
Условия работы .?.
Темп-ра
Фон
метод добавок. Для опр-ия конц. дост-но снять 2 полярограммы :1- анализ р-р; 2- анализ р-р +доб-ки. Берем фиксир-ем V и снимаем завис-ть затем доб-ем доб-ку:
Хроматографический метод анализа. Теоретические основы метода
Хроматография - метод разделения и определения вещества, основанный на распределении компонентов между двумя фазами - подвижной и неподвижной.
Хроматография - процесс, основанный на многократном повторении актов сорбции и десорбции вещества при перемещении его в потоке подвижной фазы вдоль неподвижного сорбента.
Сорбция - процесс поглощения твердым телом или жидкостью (сорбентом) газообразного или растворенного вещества (сорбата). Десорбция – процесс, обратный сорбции.
Сорбция подразделяется на адсорбцию и абсорбцию.
Адсорбция – процесс поглощения вещества (адсорбата) поверхностью твердого или жидкого адсорбентаю
Абсорбция - поглощение вещества всем объемом абсорбента.
Хемосорбция - поглощение вещества сорбентом, сопровождающееся образованием химических связей (соединений) сорбируемого компонента с сорбентом.
Вещество подвижной фазы (носитель и разделяемая проба) непрерывно вступает в контакт с новыми участками сорбента и частично сорбируется, а сорбированное вещество контактирует с новыми порциями подвижной фазы и частично десорбируется. При постоянной температуре адсорбция увеличивается с ростом концентрации раствора или давления газа.
Зависимость количества поглощенного вещества от концентрации раствора или давления газа при постоянной температуре называют изотермой адсорбции . Математическая зависимость количества поглощенного вещества (адсорбция) от концентрации поглощаемого вещества (равновесной) выражается уравнением Лэнгмюра: n = n∞ (bc/(1+bc)),
где n – количество адсорбированного вещества при равновесии; n∞ - максимальное количество вещества, которое может быть адсорбировано на данном адсорбенте; b – постоянная, с – равновесная концентрация.
П о
Лэнгмюру на поверхности твердого тела
имеется некоторое число мест с минимальной
энергией, расположенных через определенные
интервалы по всей поверхности. Их число
равно n∞ .
о
Лэнгмюру на поверхности твердого тела
имеется некоторое число мест с минимальной
энергией, расположенных через определенные
интервалы по всей поверхности. Их число
равно n∞ .
На этих местах могут адсорбироваться молекулы (ионы) из раствора или газа.
В области небольших концентраций изотерма линейна. При bc << 1 знаменатель становится равным 1, и уравнение принимает линейный вид: n = n∞ bc = Гс - уравнение Генри, Г – коэффициент Генри.
При высокой концентрации bc >> 1 уравнение Лэнгмюра принимает вид: n = n∞,
Известны более сложные случаи зависимости адсорбированного вещества от концентрации раствора или давления газа: изотерма адсорбции может быть вогнутой или S-образной.
Это может быть вызвано образованием на поверхности адсорбента не моно- , а полимолекулярного слоя, а также неоднородностью поверхности твердых тел и другими причинами.
При адсорбции двух или нескольких разных веществ уравнение адсорбции для i – го компонента принимает более сложный вид: n = n∞ (bc/(1+∑bici))
Кол-во адсорб-го в-ва определяется не только его концентрацией, но но и сродством к адсорбенту.
При адсорбции нескольких веществ проявление сродства к адсорбенту особенно заметно, так как возможно вытеснение одних сорбированных веществ другими, обладающими большим сродством, хотя имеющими, в ряде случаев меньшую концентрацию.
Классификация хроматографических методов анализа.
Неподвижной (стационарной) фазой служит твердое пористое вещество (сорбент) или пленка жидкости, нанесенная на твердое вещество.
Подвижная фаза - жидкость или газ, протекающая (протекающий) через неподвижную фазу, иногда под давлением.
Классификация: По агрегатному состоянию:
По способу относительного перемещения фаз.
 а)
Фронтальная хроматография
– простейший по методу вариант
а)
Фронтальная хроматография
– простейший по методу вариант
Через колонку с адсорбентом непрерывно пропускают анализируемую смесь: компоненты В + С и растворитель А. В растворе, вытекающем из колонки, определяют концентрацию каждого компонента и строят график в координатах: концентрация вещества – объем раствора, прошедшего через колонку. Это и есть хроматографическая или выходная кривая. Из-за сорбции веществ В и С сначала из колонки будет вытекать растворитель А, затем растворитель и менее сорбирующийся компонент В, а потом появится и С. Через некоторое время состав раствора при прохождении через колонку меняться не будет. Метод используется редко, в основном для очистки раствора от примесей.
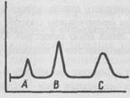 б)
Проявительный (элюентный) метод.
б)
Проявительный (элюентный) метод.
Анализируемую смесь (А + В + С) вводят в колонку в виде порции раствора или газа. Колонку непрерывно промывают газом –носителем или растворителем. При этом компоненты анализируемой смеси разделяются на зоны: хорошо сорбирующееся вещество С занимает верхнюю часть колонки, менее сорбирующиеся вещества В и А - соответствующие более низкие зоны. В газе или растворе вытекающем из колонки, сначала появляется компонент А (наименее сорбируемый), затем чистый растворитель, далее компонент В, затем чистый растворитель, и последним выходит компонент С (наиболее сорбируемый). Чем больше концентрация компонента, тем выше пик и больше площадь под пиком. Метод наиболее часто применяется на практике и дает возможность разделить сложные смеси.
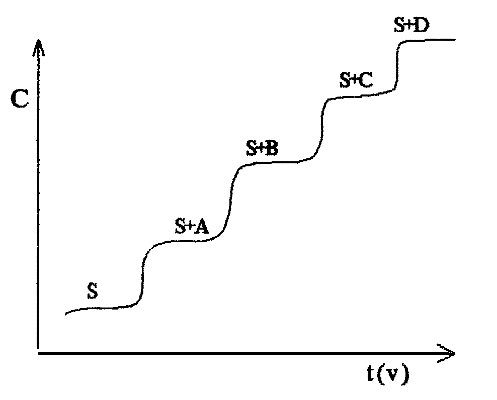 в)
вытесительный метод
в)
вытесительный метод
Анализир смесь комп-тов АВС в раств-ле S вводят в колонку и промывают р-ром в-ва D (вытеснитель), кот сорбир-ся лучше, чем любой из комп-тов анализ смеси. Конц-ия р-ра при этом не уменьшается в отличие от проявительного метода.
Недостаток в том, что накладываются зоны одного в-ва на зону другого, тк зоны комп-тов не разделены зоной растворителя.
Методика проведения
Выбирают нужные фазы – подвижную и неподвижную - в зависимости от свойств анализируемых веществ. Устанавливают необходимый режим хроматографа: →скорость подачи подвижной фазы
→температуру;
→ детектор.
Проводят хроматографическое разделение и регистрируют сигнал.Регистрируют хроматограмму - зависимость сигнала прибора (ось ОУ) от времени (ось ОХ). Анализируя и обрабатывая хроматограмму, определяют содержание каждого компонента в подвижной фазе после ее выхода из колонки. Хроматограммы получают с помощью хроматографов, основными составляющими которых являются хроматографическая колонка и детектор. Детектор - прибор проточного типа, который непрерывно фиксирует какое-либо свойство вещества на выходе из колонки.
Бумажная и тонкослойная хроматографии.
Это вариант хроматографии в плоскости (на бумаге). Его достоинства: простота, наглядность, экспрессность, возможность анализа микрообъектов.
Разделение веществ происходит вследствие распределения их между водной фазой, содержащейся в целлюлозе, и любой другой подвижной фазой. Бумага - неподвижная жидкая фаза
На один из концов этой полоски наносят каплю анализируемого раствора. Затем этот конец бумаги опускают в ванночку с растворителем. При медленном продвижении растворителя через поры бумаги компоненты раствора непрерывно перераспределяются между двумя жидкими фазами.
Если различные компоненты анализируемого раствора имеют неодинаковые коэффициенты распределения, то скорость продвижения отдельных компонентов будет различна. В результате одни компоненты движутся быстрее вслед за фронтом поднимающегося растворителя, другие медленнее, а некоторые остаются на стартовой линии. После разделения бумажную хроматограмму высушивают и проявляют, опрыскивая из пульверизатора раствором вещества, дающего цветные реакции с компонентами исследуемого раствора.
Для оценки хроматографического поведения веществ используют величину используют величину Rf = l/L, которая равна отношению расстояния l , пройденного веществом, к расстоянию L, пройденному растворителем.
Разделение веществ практически возможно, если: Rf2 - Rf1 ≥ 0,1 и Rf3 - Rf2 ≥ 0,1 .
Бумажные хроматограммы можно получать путем восходящего, нисходящего, горизонтального и радиального движения растворителя
Преимещества: возможностьвыполнения метода при минимальном лабораторном оборудовании, простая методика, высокая экономичность, высокая селективность и чувствительность.
Недостаток метода: сильное размывание хроматографических зон, связанное с неоднородностью бумаги, поэтому берут достаточно длинный кусок бумаги, что приводит к увеличению длительности и большому расходу растворителя.
Тонкослойная хроматография (ТСХ)
ТСХ: тонкий слой сорбента прочно связан с подложкой из стекла, пластмассы, алюминиевой фольги.
Подвижная фаза и компоненты анализируемой смеси с различными скоростями перемещаются по тонкому слою сорбента в одном направлении под действием капиллярных сил. При этом компоненты смеси разделяются и располагаются отдельными зонами в соответствии с коэффициентами распределения в данной хроматографической системе
Подвижная фаза: смеси органических растворителей, водных растворов кислот, солей, комплексообразователей…
Положение зон на хроматограмме также как и в бумажной хроматографии хар-тся величиной Rf
Достоинсва: экспрессность, доступность, дешевое оборудование, наглядность и иныормативность
Недостатки: трудоемкость и использование токсичных раств-лей
Газовая хроматография. Колонки и детекторы. Эффективность колонки
Газовая хроматография – процесс разделения компонентов смеси, основанный на различии в равновесном распределении компонентов между двумя фазами – газом-носителем (подвижная фаза ПФ) и либо твердой фазой (-газоадсорбционная хроматография), либо жидкостью, нанесенной в виде тонкой пленки на поверхность твердого носителя или стенки хроматографической колонки (неподвижная фаза НФ)(- газожидкостная хроматография – ГЖХ.)
ГЖХ – наиболее распространненый метод газовой хроматографии.
Анализируемая смесь (обычно раствор) летучих компонентов переводится в парообразное состояние и смешивается с потоком инертного газа-носителя, образуя с ним подвижную фазу. Эта смесь проталкивается новой порцией непрерывно подаваемого газа-носителя и попадает в хроматографическую колонку, заполненную неподвижной жидкой фазой. Разделяемые компоненты распределяются между ПФ и НФ в соответствии с их коэффициентами распределения: К = С(НФ)/С(ПФ).
Поток газа-носителя увлекает с собой разделяемую парообразную смесь вдоль хроматографической колонки, в результате чего процесс сорбции-десорбции повторяется многократно.
Переходы разделяемых веществ из ПФ в НФ и обратно совершаются по всей длине колонки, затем пары разделяемых веществ покидают колонку вместе с газом-носителем. Так как сродство различных разделяемых веществ к НФ различно, то в процессе сорбционно-десорбционных переходов они задерживаются в НФ неодинаковое время. Чем выше температура кипения и относительная растворимость вещества в НФ, тем дольше оно находится в НФ, тем позже покидает хроматографическую колонку.
Из хроматографической колонки вместе с газом-носителем выходят зоны (объемы) парообразных хроматографируемых веществ.Пары разделенных компонентов вместе с газом-носителем поступают в детектор, генерирующий эл-ий сигнал. Чем больше концентрация компонента, тем больше сигнал
С ростом температуры в колонке увеличивается средняя скорость движения молекул в парогазовой фазе, в результате этого уменьшается разность скоростей между «убегающими» и «отстающими» частицами одного и того же компонента. Зоны разделяемых веществ (пики) становятся более узкими, менее размытыми. Но снижается селективность.
При понижении темп-ры увелич-ся длительность анализа, но при этом улучшается степень разделения.
Колонки в газовой хроматографии - металлические или стеклянные трубки (прямые, изогнутые, спиральные) с внутренним диаметром 0,1 – 5 мм и длиной до нескольких метров.
Существует два типа колонок:
наполненные или насадочные: это металлические или стеклянные колонки, изогнутые в виде спирали с диаметром 1,5 -5 мм, заполненные насадкой; насадка – частицы с твердой основой, на поверхность которой нанесен слой жидкой неподвижной фазы;
капиллярные колонки – изготовленные из кварцевого стекла трубки, внутренняя поверхность которых покрыта тонкой пленкой жидкой неподвижной фазы; роль твердого носителя играет внутренняя поверхность самой колонки; длина такой колонки от нескольких десятков до нескольких сотен метров, внутренний диаметр – 0,1 – 0,6 мм.
Капиллярные колонки более эффективны для разделения многокомпонентных смесей.
Условиями эффективного разделения являются
достаточно большое число теоретических тарелок колонки ( у капиллярных от 100 000 и более)
высокая селективность неподвижной фазы;
достаточная ёмкость неподвижной фазы.
Детекторы
Детектор нужен для обнаружения изменений в составе газа или р-ра (ПФ), прошедшего через колонку.
Показания детектора преобразуются в электрический сигнал и передаются фиксирующему или записывающему прибору (например, на электронный потенциометр).
Основные характеристики детектора: чувствительность, пределы детектирования, инерционность и диапазон линейной зависимости между концентрацией и величиной сигнала.
Детекторы подразделяются на
дифференциальные, отражающие мгновенное изменение концентрации;
интегральные, суммирующие изменение концентрации за некоторый отрезок времени; недостатки (инерционность и низкая чувствительность).
Детектор выбирают в завис-ти от свойств изучаемой системы, агр-го состояния фаз и других особенностей.
Катарометр –универсальный. Основан на сравнении теплопроводности двух газовых потоков: газа-носителя и смеси газа-носителя с анализируемым компонентом. При анализе проба не разрушается
Пламенно-ионизационный – ПИД – более чувствителен. В основе работы - увеличение электропроводности водородного пламени, помещенного в электрическое поле в присутствии органических веществ. Компоненты пробы после детектирования полностью разрушаются.
Детектор электронного захвата (ДЭЗ) : газ, выходящий из колонки, в специальной ячейке с двумя электродами непрерывно облучается потокам β-частиц
Чувствителен только к некоторым органическим соединениям .
Хроматографические параметры.
Время удерживания
Время от момента ввода анализируемой пробы до момента регистрации максимума хроматографического пика tR – время удерживания. Время удерживания складывается из времени пребывания вещества в подвижной фазе tm и времени пребывания в неподвижной фазе tn: tR = tm + tn.
Значение tm фактически равно времени прохождения через хроматограф несорбируемого компонента – подвижной фазы: tm = L/F, где L- длина колонки, F – линейная скорость движения потока (элюента).
Время удерживания tR не зависит от количества пробы, вводимой в колонку, но зависит от природы вещества и природы сорбента (если изотерма сорбции линейна). При переходе на другую колонку, время удерживания может меняться.
Для характеристики истинной удерживающей способности колонки вводят исправленное время удерживания: tꞌR = tR – tm.
Удерживаемый объем
VR - объем подвижной фазы, который нужно пропустить через колонку с определенной скоростью, чтобы элюировать вещество: VR = F∙tR ,
где F – объмная скорость потока подвижной фазы, см3/с или мл/мин.
Исправленный объем удерживания VꞌR = VR - Vm
По полученной хроматограмме смеси можно рассчитать экспериментальные значения хроматографических параметров, определив tm , tR, VR, Vm и ширину хроматографического пика ῳ у основания.
Коэффициент ёмкости К
К = (VR - Vm)/Vm = (tR - tm)/tm .
Чем выше К, тем большее время находится в неподвижной фазе компонент.
Эффективность хроматографической колонки.
Под эффективностью хроматографической колонки понимают степень размывания хроматографического пика. Количественно эффективность колонки может быть выражена числом теоретических тарелок N или высотой теоретической тарелки Н.
Теоретическая тарелка - участок зоны внутри колонки, на котором устанавливается равновесное распределение данного вещества между ПФ и НФ. Н = L/N
N = 16(tR/ ῳ)2 - рассчитывают из экспериментальных данных. Значение времени удерживания tR и ширина пика ῳ должны быть выражены в одних единицах (t, длины…)
Критерий разделения
k = 2∆l/(ῳ1+ ῳ2) , l – расстояние между пиками. (k=1 – хорошее разрешение).
Разрешение
Разделение 2-х соседних пиков характеризуется разрешением (или степенью разделения) Rs:
Rs = 2(tR2 - tR1)/(ῳ1+ ῳ2) = 2∆t/(ῳ1+ ῳ2),
где tR2 , tR1 - время удерживания компонентов 2 и 1 соответственно, ῳ2 и ῳ1 – ширина их пиков. Разрешение – мера полноты разделения двух веществ. Разделение считается полным, если Rs ≥ 1,5.
Фактор разделения ( коэффициент селективности)
Разрешение зависит не только от размывания пиков. Но и от селективности неподвижной фазы, которая оценивается фактором разделения (или коэффициентом селективности) α:
α = tꞌR2/ tꞌR1 = к2/ к1, где к2 и к1 – коэффициенты распределения.
Коэффициенты распределения к = С(НФ) / С (ПФ), где С –концентрация.
Для разделения 2-х веществ необходимо подобрать условия разделения так, чтобы к1 ≠ к2 и α > 1. Разрешение зависит и от коэффициента ёмкости разделяемых компонентов, так как с ростом коэффициента ёмкости размывание хроматографического пика увеличивается.
Суммарное влияние основных параметров хроматографической колонки (эффективности, селективности и коэффициента ёмкости) на разрешение хроматографических пиков описывается уравнением:
Rs = ¼√N∙((α-1)/α)(K2/(1 + K2).
Хроматограммы. Параметры разделения. Эффективность хроматографической колонки. Качественный и количественный хроматографический анализ.
Сигнал от детектора преобразуется, усиливается и регистрируется в виде хроматограммы.
Профиль хроматограммы - совокупность данных о числе, относительном расположении, интенсивности и форме пиков, воспринимаемая как единый образ, характеризующий природу, происхождение и особенности состава анализируемой многокомпонентной системы
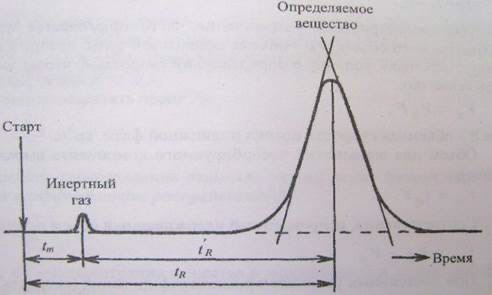
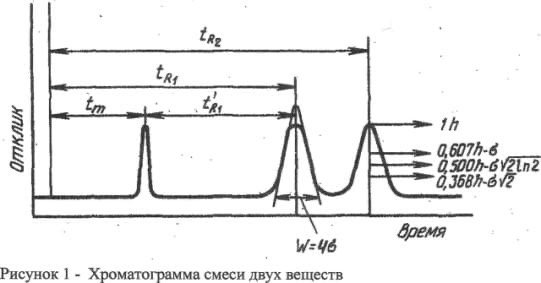 Хроматограмма
индивидуального вещества
Хроматограмма
духкомпонентной системы
Хроматограмма
индивидуального вещества
Хроматограмма
духкомпонентной системы
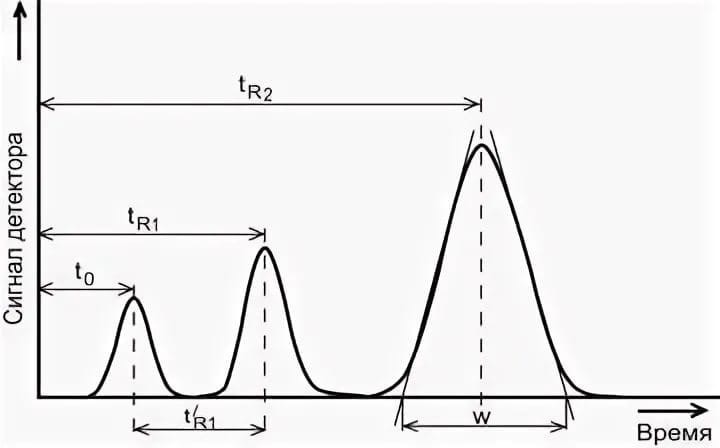
 Хроматограмма
двухкомпонентной системы Хроматограмма
многокомпонентной системы
Хроматограмма
двухкомпонентной системы Хроматограмма
многокомпонентной системы
Хроматографические параметры.