

МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ
Федеральное государственное автономное образовательное учреждение высшего профессионального образования
«Национальный исследовательский ядерный университет «МИФИ» Обнинский институт атомной энергетики Медицинский факультет
Кафедра микробиологии, вирусологии, иммунологии
Дисциплина
«Клиническая вирусология и иммунология»
Презентация на тему:
Первичные (генетически обусловленные)
иммунодефициты. 
Выполнила: студентка VI курса группы ЛД3-С13Б Соловьянова Е.А.
Проверила: к.м.н., доцент, зав. каф. Колесникова С.Г.
Обнинск, 2018 г.
Первичные иммунодефициты (ПИД)
Гетерогенная группа наследственных расстройств, причиной которых являются мутации специфических генов с дефектами в одном или нескольких компонентах иммунной системы.
Реализация генетического дефекта приводит к появлению широкого спектра заболеваний.
Эти заболевания – результат повышенной чувствительности пациентов к инфекциям, предрасположенности к аутоиммунным и онкологическим заболеваниям, а также аллергии.

Классификация.
1 группа. Комбинированные Т- и В- клеточные иммунодефициты.
1.1.Х- сцепленная тяжелая комбинированная иммунная недостаточность
1.2.Тяжелая комбинированная иммунная недостаточность с дефицитом аденозиндезаминазы
1.3.Тяжелая комбинированная иммунная недостаточность с дефицитом пуриннуклеозидфосфорилазы
1.4.Синдром Оменна
1.5.Тяжелая комбинированная иммунная недостаточность обусловленная дефицитом JAK3
1.6.Дефицит молекул главного комплекса гистосовместимости класса I
1.7.Дефицит молекул главного комплекса гистосовместимости класса II
2 группа. Преимущественные дефициты антител.
2.1.Агаммаглобулинемия с отсутствием В-клеток
2.2.Гипогаммаглобулинемия с нормальным или сниженным количеством В-клеток
2.3.Первичные В-клеточные дефекты. Синдромы, связанные с блоком переключения класса иммуноглобулинов
2.4.Селективный дефицит IgA
2.5.Другие первичные иммунодефициты, связанные с дефицитом изотипов или легких цепей иммуноглобулинов
2.6.Дефицит специфических антител с нормальной концентрацией иммуноглобулинов
2.7.Транзиторная гипогаммаглобулинемия детей раннего возраста
3 группа. Синдромы иммунодефицитов с хорошо охарактеризованными клиническими признаками.
3.1.Синдром Луи-Бар
3.2.Синдром Неймеген
3.3.Синдром Вискотта-Олдрича
3.4.Синдром Ди Джорджи
3.5.Гипер-IgE-синдром

Классификация.
4 группа. Генетические нарушения регуляции иммунитета.
4.1.Семейный гемофагоцитарный лимфогистиоцитоз
4.2.Иммунодефициты с гипопигментацией
4.3.Синдром Чедиака – Хигаси
4.4.Х-сцепленный лимфопролиферативный синдром
4.5.Аутоиммунный лимфопролиферативный синдром
4.6.Х-сцепленные иммунодисрегуляция, полиэндокринопатия, энтеропатия
5 группа. Врожденные дефекты фагоцитов.
5.1.Тяжелые врожденные нейтропении
5.2.Циклическая нейтропения
5.3.Дефициты адгезии лейкоцитов
5.4.Хроническая гранулематозная болезнь
5.5.Дефицит глюкозо-6-фосфатдегидрогеназы нейтрофилов
5.6.Дефицит миелопероксидазы
6 группа. Дефекты врожденного иммунитета: рецепторов и сигнальных компонентов.
7 группа. Аутовоспалительные нарушения.
8 группа. Дефициты комплемента.
8.1. Наследственный ангионевротический отек

“Настораживающие признаки”
1.Частый отит (6-8 раз в год и чаще)
2.Синусит (4-6 раз в год)
3.Пневмония (2 раза в год и чаще)
4.Абсцессы кожи и внутренних органов (особенно повторные)
5.Антибиотикотерапия для купирования инфекции с применением а/б в/м или per os в течение 2 мес и более, а также
сисползованием в/в а/б
6.Не менее двух перенесенных инфекций таких, как –
менингит, сепсис, остеомиелит
7.Отставание ребенка в росте и массе тела (по возрастным нормам)
8.Персистирующая молочница или грибковые поражения кожи в возрасте старше 1 года
9.Указание на наличие в семье больных ПИД, ранние смерти детей от тяжелых инфекций

Врожденные дефекты фагоцитов.
Врожденная форма иммунной недостаточности с дефектами числа и функций фагоцитов, в частности нарушением подвижности, хемотаксиса, адгезии, эндоцитоза, киллинга, деградации фагоцитированных частиц, секреции цитокинов и др.

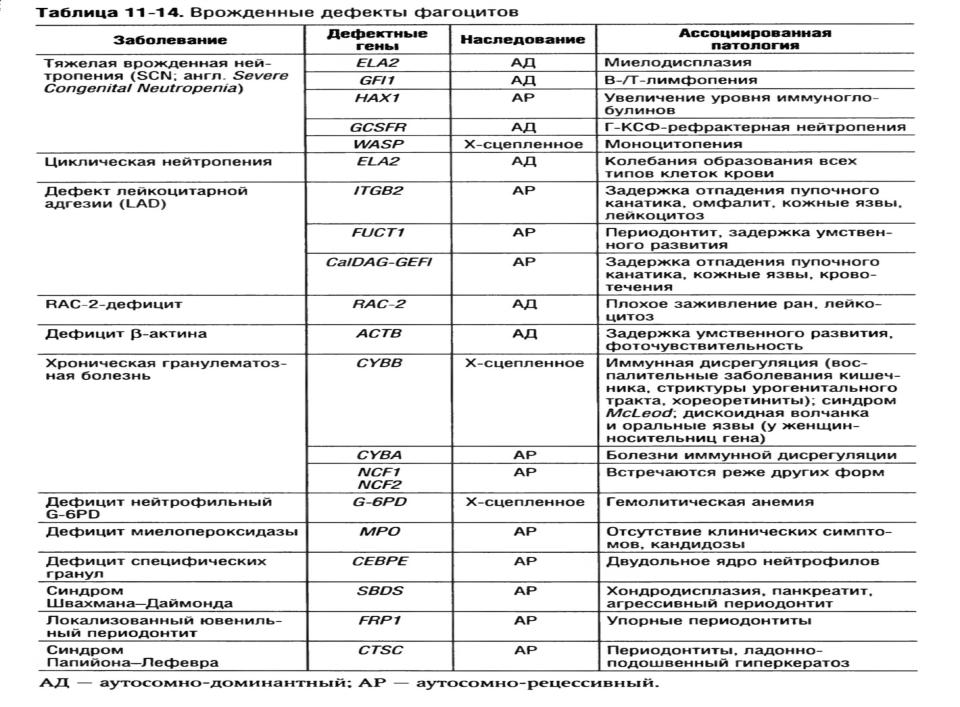

1. Тяжелые врожденные нейтропении.
•Характеризуется ранними проявлениями бактериальных инфекций и персистирующей тяжелой нейтропенией.
•Механизм развития: мультигенные нарушения с различными формами наследования.
•Клиника: рецидивирующие бактериальные инфекции, манифестация около 1 года. Частые проявления – неглубокие абсцессы, язвы в полости рта, инфекц поражения кожи, омфалит, пневмония, средние отиты, неврологические нарушения.
•Диагностика: нейтропения, связанная с рецидивир инфекц заболеваниями, диагностический критерий – содержание нейтрофилов меньше 500/мм3 +
одновременное повышение чувствительности к рецидивирующим бактериальным инфекциям, начиная с раннего возраста. При исследовании костного мозга выявляют нарушенное созревание нейтрофилов из предшественников в ранней стадии (дифференцировка промиелоцитов и миелоцитов), клеточность обычно нормальная или слегка снижена, тогда как кол-во эозинофилов и моноцитов увеличено.
•Лечение: трансплантация костного мозга пациентам до 2-летнего возраста. Пациентам, которым не требуется пересадка костного мозга рекомендован рекомбинантный Г-КСФ
(гранулоцитарный колониестимулирующий фактор), улучшает прогноз и качество жизни пациентов, 90% положительно отвечают на лечение увеличением числа
нейтрофилов и снижением числа инфекционных заболеваний. Но! У пациентов с мутацией гена, кодирующего рецептор Г-КСФ, которые не отвечают на лечение Г-КСФ, продолжаются тяжелые бактериальные инфекции, развивается миелодисплазия. В этом случае требуется пересадка стволовых клеток.

2.Дефициты адгезии лимфоцитов.
•Характеризуется кожными язвами, плохо заживающими ранами и рецидивирующими бактериальными инфекциями.
•У пациентов выявляют аномалии рецепторов необратимой адгезии CD11/CD18 (интегрины). Если на клетке экспрессируется меньше 1% адгезивных молекул, развиваются угрожающие жизни инфекционные процессы. Если до 10%, то возможно развитие септицемии, гингивита, периодонтита, некротических поражений кожи, кишечных или перианальных свищей.
•Механизм развития: в норме при воспалении происходит накопление лейкоцитов в зоне поражения (трансмиграция этих клеток из крови через эндотелиальный барьер). Основная проблема лейкоцитарной адгезии – лейкоциты не могут покинуть кровеносные сосуды и мигрировать в зону воспаления.
•При отсутствии адекватного лечения более 75% пациентов умирают до 2х летнего возраста.
•Диагностика: сочетание ранних бактериальных инфекций с резко выраженной нейтрофилией (до 50000-100000/мкл) при инфицировании патогеном и в пределах 15000/мкл в отсутствии инфекции. Так же подтверждается диагноз экспрессией молекул CD18, CD15a на нейтрофилах. В крови выявляют
массивный нейтрофильный лейкоцитоз до 15000-75000, и даже 100000 клеток в 1 мкл, что объясняется неспособностью этих клеток выйти на периферию из сосудистого русла.
•Лечение: антибиотикотерапия, рекомбинантный Г-КСФ (гранулоцитарный колониестимулирующий фактор), а также стволовые клетки.В будущем возможна генная терапия, методы которой разрабатываются.