

5 quants / Лабораторный практикум Квантово-химическое моделирование соединений в пакете HyperChem Учебно-методическое пособие
.pdf6.1. Вывод характеристик и установка параметров атомов
Для того чтобы получить информацию об основных характеристиках атома его нужно выделить. Перед выделением инструментом удостоверитесь, что в меню Select, выбрана опция Atoms, а не Multiple Selections. В строке состояния появляются: номер атома, тип, заряд для выбранного силового поля молекулярной механики или эффективный заряд, рассчитанный квантово-химическим методом. Она также показывает x, y и z-координаты атома (рис. 1.36).
Пункты меню Build позволяют установить необходимый Вам для расчета тип атома (Set Atom Type), заряд (Set Charge), массу (Set Mass) и ограниченную геометрию (Constrain Geometry) (рис. 1.37, 1.38), отличные от тех, которые устанавливаются программой по умолчанию.
Рис. 1.36. Свойства выбранного атома кислорода в строке состояния
Рис. 1.37. Меню ручного назначения |
Рис. 1.38. Установка заряда для |
свойств для выбранного атома |
атома кислорода |
31
6.2. Измерение длины связи
Набор параметров, достаточных для создания структурной молекулярной модели из N атомов, состоит из длин валентных связей (N-1), валентных (N-2) и торсионных углов (N-3). Всего 3N-6 параметров молекулярной геометрии.
HyperChem имеет библиотеку длин связей между атомами конкретного типа и гибридизации, что устанавливается по умолчанию построителем модели (Model Builder). Когда информация о длине связи в библиотеке отсутствует, то HyperChem использует среднее значение ковалентных радиусов двух атомов.
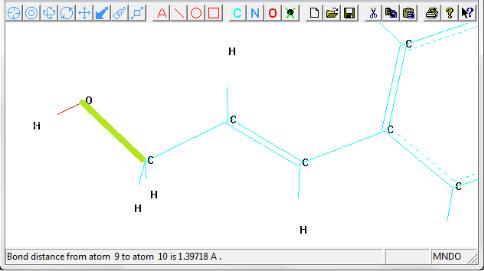
Для измерения расстояния нужно изменить текущий инструмент
на  (Select) и выделить эту связь. Перед выделением удостоверитесь, что в меню Select, выбраны опции Atoms и Multiple Selections (Множественные выборы). В строке состояния появляется значение длины связи между двумя атомами, выраженное в ангст-
(Select) и выделить эту связь. Перед выделением удостоверитесь, что в меню Select, выбраны опции Atoms и Multiple Selections (Множественные выборы). В строке состояния появляется значение длины связи между двумя атомами, выраженное в ангст-
ремах (Å) (рис. 1.39).
Рис. 1.39. Измеренное расстояние между соседними атомами О и С
Длина связи – расстояние в равновесном состоянии между положениями ядер атомов, которые образовали данную связь.
При этом в меню Build становиться активным пункт Constrain bond length, который позволяет Вам установить желаемую длину связи, отличную от той, которую устанавливает Построитель моделей программы по умолчанию.
32
Прежде, чем Вы приступите к измерению расстояния между двумя несвязанными атомами, нужно в меню Select обязательно отметить пункт Multiple Selections. Это позволяет одновременно выделить более одного атома в рабочем окне. L-щелчком инструментом
 выделите два любых (не соседних) атома, при этом строка состояния показывает расстояние между ними.
выделите два любых (не соседних) атома, при этом строка состояния показывает расстояние между ними.
6.3. Измерение углов между связями
Валентный угол — угол, образованный направлениями химических связей, исходящими из одного атома. Знание валентных углов необходимо для определения геометрии молекул. Валентные углы зависят как от индивидуальных особенностей присоединенных атомов, так и от гибридизации атомных орбиталей центрального атома.
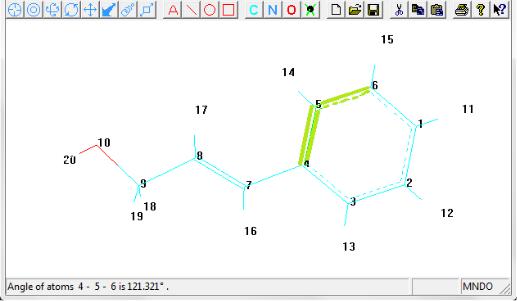
Для того чтобы измерить угол между двумя связями нужно последовательно выделить одинарными щелчками первый, второй и третий атомы, связи между которыми и образуют угол. При этом второй атом должен находиться в вершине этого угла. Величина угла в градусах появится в строке состояния (рис. 1.40).
Рис. 1.40. Измерение валентного угла внутри бензольного кольца
При этом более удобным является отображение подписей атомов в виде номеров (Numbers), т.к. это позволяет легко отличить в молекулы атомы одного и того же элемента друг от друга.
33

Существует второй способ выделение угла между связями. Сделайте L-перемещение от атома углерода №4 сразу к атому углерода №6 (т.е. от одного концевого атома к другому концевому атому, образующих валентный угол), и только потом отпустить кнопку
(рис. 1.41).
HyperChem имеет библиотеку валентных углов между связями, образованными атомами конкретного типа и гибридизации, что устанавливается по умолчанию Построителем модели (Model Builder). Так, для тетраэдрической структуры выставляется угол 109 градусов, для тригональных - 120 градусов, а линейные структуры имеют угол 180 градусов. После выделения связи в меню Build становится активным пункт Constrain Bond Angle, что позволяет Вам задать величину данного угла вручную.
Рис. 1.41. Альтернативный способ выделения валентного угла
6.4. Измерение торсионных углов
Торсионный угол – стереохимический элемент молекулярной структуры, двугранный угол между плоскостями, образованными атомами фрагмента молекулы.
Последовательно выделите первый, второй (вершина угла) и третий атомы в одной плоскости, а далее четвертый атом, который расположен вне плоскости первых трех атомов молекулы. В строке стояния появиться величина торсионного угла между плоскостями, в которых лежат три первых атома, и плоскостью четвертого атома (рис. 1.42). Альтернативным способом является соединение L- протяжкой сразу первого и четвертого атома торсионного угла.
34

После выделения торсионного угла пункт Constrain Bond Torsion в меню Build становится активным, что позволяет Вам изменить по желанию эту величину.
Рис. 1.42. Измерение торсионного угла между CH-связями алифатической цепочки
6.5. Отображение водородных связей
Если молекулярная геометрия благоприятна для образования водородных связей, HyperChem вычисляет их и выводит на дисплей (рис. 1.43). Водородные связи формируются, если расстояние до водородного донора - менее чем 3,2 Å и угол ковалентной связи донора и акцептора - менее чем 120 градусов.
Рис. 1.43. Изображение водородных связей между двумя молекулами глицина
Чтобы подтвердить условия для показа водородной связи:
35
1.В меню Display отметьте пункт Show Hydrogen Bonds (Пока-
зать водородные связи).
2.Там же выберите Recompute H Bonds (Пересчитать водо-
родные связи).
HyperChem отображает водородные связи пунктирной линией. Водородные связи не вычисляются автоматически в каждой конфигурации, поэтому, когда Вы изменяете геометрию молекулы, водородные связи приходится вычислять заново.
7. Методы расчета в пакете HyperChem
Перед проведением расчета в HyperChem при помощи команд из меню Compute необходимо выбрать и настроить метод расчета энергии систем и равновесной геометрии.
Меню Setup (Установка) содержит опции для проведения моле- кулярно-механических и квантово-химических расчетов энергетических, геометрических и электронных параметров молекулярных систем (табл. 1.2). Программа HyperChem может выполнять расчеты методами: молекулярной механики с использованием четырех модельных потенциалов (MM+, AMBER, BIO+ и OPLS), полуэмпирическими квантово-химическими методами (12 вариантов, от расширенного метода Хюккеля, CNDO, INDO, MINDO3, MNDO, AM1, PM3, и ZINDO/S до TNDO), неэмпирический метод ХартриФока (ab initio) и метод функционала плотности (DFT) в различных базисах.
Таблица 1.2. Содержимое меню Setup
Molecular |
Использование ньютоновской механики для расчетов моле- |
mechanics |
кулярных потенциалов методами молекулярной механики |
Semi- |
Использование одного из полуэмпирических квантово- |
empirical |
химических методов расчета параметров молекул |
Ab Initio |
Выбор Ab Initio позволит использовать неэмпирический ме- |
|
тод Хартри-Фока |
Density |
Выбор неэмпирического метода функционала плотности |
Functional |
|
Periodic |
Периодический ящик. Выбор этого пункта позволит помес- |
box |
тить молекулярную систему в периодический куб, содер- |
|
жащий молекулы воды. Вычисления в периодическом кубе |
|
проводятся только молекулярно-механическими методами |
|
или методами молекулярной динамики |
36
Таблица 1.2. Продолжение
Restraints |
Ограничения. Добавление граничных условий (действующих |
|
сил) для 1, 2, 3 и 4 выделенных типов атомов для молекуляр- |
|
ной механики и квантовой химии. Этот пункт меню оста- |
|
ется неактивным, если такого выделения сделано не было |
Set velocity |
Присвоение скорости. Выбор этого пункта позволяет уста- |
|
навливать скорости атомам, входящим в систему. Этот |
|
пункт используется в некоторых видах молекулярно- |
|
динамических расчетов |
Set Finite |
Установить ограниченное поле. Установка параметров |
Field |
внешнего ограниченного электрического поля |
Network |
Сетевое вычисление. Выбор этого пункта позволяет прово- |
|
дить расчеты с использованием удаленного сервера |
Edit |
Редактирование параметров. Просмотр и редактирование |
parameters |
используемых параметров вычислений |
Select |
Выбор набора параметров. Этот пункт позволяет устанав- |
parameter |
ливать альтернативный набор параметров для молекуляр- |
set |
ной механики |
Compile |
Компиляция файла параметров. Преобразование нового |
parameter |
файла параметров из текстового вида или файла базы дан- |
file |
ных в двоичный код, используемый программой HyperChem |
Reaction |
Карта реакции. Выбор этого пункта позволяет строить |
map |
карту реакции, т.е. путь от реагентов к продуктам реакции |
|
и синхронного поиска переходного состояния. Этот пункт |
|
остается неактивным до тех пор, пока не сделан выбор реа- |
|
гентов и продуктов реакции |
После того, как Вы выбрали нужную опцию в меню Setup, используйте меню Compute, для проведения расчетов необходимых характеристик.
7.1.Эмпирические методы расчета
7.1.1.Методы молекулярной механики
Выбор в меню Setup пункта Molecular Mechanics (молекулярная механика) (рис. 1.43) позволяет использовать классический ньютоновский метод для нахождения энергии химического соединения в одной точке, равновесной геометрии молекулы и молекулярной динамики объектов. При этом не используется квантовомеханический подход, как в полуэмпирических методах.
В методе молекулярной механики атомы рассматриваются как ньютоновские частицы определенной массы, которые взаимодейст-
37
вуют друг с другом посредством эмпирически задаваемых потенциальных полей. Потенциальная энергия взаимодействия зависит от взаимодействующих атомов, длины и углов валентных связей, торсионных углов и нековалентных взаимодействий (сил Ван-дер- Ваальса, электростатических взаимодействий и водородных связей). Вклад в молекулярную энергию также включает в себя упругую энергию связи, описываемую законом Гука, энергию изгиба валентных углов и угловых деформаций. В этих расчетах силы, действующие на атомы, представляются в виде функций координат атомов и силовых постоянных, т.е. коэффициентов жесткости упругих сил, связывающие пары атомов.
Методы молекулярной механики позволяют минимизировать энергию очень больших многотысячных молекулярных систем, при разумных вычислительных и временных затратах. Эти методы могут служить моделью для оценки потенциальной энергии молекулы с учетом всех степень свободы. Но они совершенно не применимы для моделирования тех систем, свойства которых определяются электронными эффектами, орбитальным взаимодействием и т.п.
Примечание: Если в рабочей области выделена только часть системы, в этом случае в расчет будут включаться взаимодействия только выделенной части. При оптимизации геометрии и расчетах методом молекулярной динамики, только атомы выделенной части будут менять свое положение в пространстве, тогда как невыделенные – нет, при этом в расчетах будут учитываться потенциальные взаимодействия между частями системы.
Для начала расчетов методом молекулярной механики в диалоговом окне необходимо выбрать метод Force fild (Силовое поле) – вид потенциальной функции для расчетов. Доступны четыре метода (MM+, AMBER, BIO+, OPLS), ссылки на которые находятся в диалоговом окне (рис. 1.43).
Метод MM+ разрабатывался для органических молекул. Он учитывает потенциальные поля, формируемые всеми атомами рассчитываемой системы, и позволяет гибко модифицировать параметры расчета в зависимости от конкретной задачи, что делает его, с одной стороны, наиболее общим, а с другой стороны, он резко увеличивает необходимые ресурсы по сравнению с остальными методами молекулярной механики. Ряд возможностей для изменения па-
38
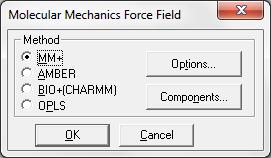
раметров этого метода можно получить, выбрав кнопку Options в окне выбора метода Силового поля (Force field).
Рис. 1.43. Выбор метода из группы Молекулярная механика
Метод AMBER разрабатывался для белков и нуклеиновых кислот. В нем существует возможность выбрать опцию либо учета всех атомов по отдельности, либо опцию объединенного атома, под которым подразумевается группа эквивалентных атомов с одинаковыми свойствами. В последнем случае несколько атомов или их группа обрабатываются как один атом с одним типом.
BIO+ – разрабатывался для биологических макромолекул и во многом повторяет AMBER.
OPLS – разработан для белков и нуклеиновых кислот. Он подобен AMBER, но более точно обрабатывает нековалентные взаимодействия.
7.1.2. Опции силового поля ММ+
Диалоговое окно ММ+ Options содержит набор следующих настроек для соответствующего силового поля (рис. 1.44): Electrostatics (электростатика) – способ расчета нековалентных электростатических взаимодействий: с использованием взаимодействий дипольного типа или частичных атомных зарядов.
•Bond dipoles (связи диполей) – используется для расчетов нековалентных электростатических взаимодействий. Значение этого параметра определяется в файле параметров MM+.
•Atomic charges (атомные заряды) – используется для расчетов нековалентных электростатических взаимодействий. Можно задавать неполные (частичные) атомные заряды посредством меню Build, пункта Set Charge, или можно использовать результаты полуэмпирических или ab initio расчетов, в рамках
39

которых рассчитываются частичные заряды для каждого атома методом Малликена.
Рис. 1.44. Диалоговое окно параметров метода молекулярной механики MM+
Cutoffs (ограничение) – данный параметр определяет максимальное расстояние для нековалентных взаимодействий, т.е. задает ограничения по расстоянию для выполнения расчетов. Для Periodic Box условия ограничений вычислений устанавливаются автоматически.
•None (ничего) – этот параметр устанавливается для расчета свойств систем в вакууме. Вычисляются все нековалентные взаимодействия.
•Switched – вводит сглаживающую функцию при расчетах нековалентных взаимодействий молекул.
•Shifted – вводит сглаживающую функцию, которая действует
на все пространство от 0 до внешней сферы. Эта функция позволяет плавно уменьшать нековалентные взаимодействия.
9Outer radius – для параметров Switched и Shifted определя-
ет максимальное расстояние, на котором нековалентные взаимодействия становятся равными 0. Обычно, это значение выбирается не менее чем на 4 Å больше, чем внутренний радиус. Для периодических граничных условий это значение равно половине минимального размера периодического ящика.
9Inner radius – выбирается только в случае установки Switched cutoffs. Это максимальное межатомное расстояние для полного учета нековалентных взаимодействий. В случае
40